
| IMGT Web resources |
|
| Here you are: IMGT Web resources > IMGT Education > Tutorials > Cancer |
Dérégulation moléculaire et cancer: prolifération, survie et inhibition de l'apoptose
Géraldine GUASCH
U119 INSERM Laboratoire d'Oncologie Moléculaire
27 Bd Leï Roure 13009 Marseille
SOMMAIRE
1. Autosuffisance en signaux de croissance
1-1. Les facteurs de croissance hématopoïétiques et les cytokines
1-2. Comment les cellules cancéreuses acquièrent-elles l’indépendance en signaux de croissance ?
2-1. Les principales voies moléculaires dans l’induction de l’apoptose
2-1-1. L’apoptose induite par les ‘’récepteurs de mort’’, TNFR1 et FAS/APO-1/CD95
2-1-2. L’apoptose induite par la mitochondrie
2-2. Les voies impliquées dans la survie cellulaire et leur surexpression dans certains cancers
2-2-1. BCL2 (B cell leukemia)
2-2-2. La voie PI3Kinase/AKT
3. Conclusions
4. Références bibliographiques
Les cellules néoplasiques, bénignes ou malignes, sont diverses et hétérogènes. Cependant, toutes ces cellules ont comme caractéristique commune de proliférer en dépit des contrôles normaux. Les cellules néoplasiques malignes ont en plus la capacité d'envahir et de coloniser les tissus environnants. Il est clairement établi que le développement d'un cancer est le résultat d'une combinaison entre, d'une part, l'activation de voies favorisant la prolifération cellulaire et, d'autre part, l'inhibition de signaux restreignant le potentiel prolifératif des cellules. Hanahan et al., (2000) ont proposé de classer ces différentes modifications en six catégories, qui semblent généralement retrouvées dans chaque type de cancer: une autosuffisance en signaux de croissance, une résistance à l'apoptose, une insensibilité aux signaux d'inhibition de croissance, un potentiel réplicatif infini ("immortalisation"), un potentiel de néovascularisation et une capacité d'invasion tissulaire. Je ne détaillerai que les deux premiers processus qui résultent d'une dérégulation dans le contrôle de la prolifération, de la survie cellulaire et de l'apoptose.
1. Autosuffisance en signaux de croissance
Une cellule normale nécessite des facteurs de croissance mitogéniques afin de passer d'un état quiescent à un état prolifératif actif. Au niveau du système hématopoïétique il existe des contrôles précis de la prolifération, de la migration, de la différenciation et de l'apoptose, via des interactions entre les progéniteurs, les cellules du stroma médullaire (ou micro-environnement) et des facteurs de croissance hématopoïétiques (Alexander et al., 1998). Aucune cellule normale n'est capable de proliférer en l'absence de ces signaux de survie et de croissance.
1-1. Les facteurs de croissance hématopoïétiques et les cytokines:
Les cytokines sont des médiateurs entre les cellules stromales, cellules essentielles du micro-environnement d'origine mésodermique (Charbord 1998), et les précurseurs hématopoïétiques. Les plus connus sont le Stem Cell Factor (SCF), les interleukines (en particulier IL-1, IL-2, IL-3, IL-6, IL-7), le ligand de FLT3, l'érythropoïétine (EPO) et les Colony-Stimulating Factors (G-CSF, M-CSF, GM-CSF). Les CSF induisent la prolifération de progéniteurs myéloïdes et induisent la formation de colonies in vitro. Ces facteurs régulateurs sont produits aussi bien par les cellules du système hématopoïétique (granulocytes, lymphocytes ou monocytes activés) que par les cellules du stroma médullaire.
Les cytokines et les facteurs de croissance régulent la croissance et la différenciation cellulaire en agissant au niveau cellulaire par l'intermédiaire de récepteurs membranaires spécifiques. L'activation d'un récepteur par son ligand entraîne sa dimérisation (homo- et/ou hétérodimérisation) et la phosphorylation de résidus tyrosine et/ou de résidus serine ou thréonine. Les récepteurs ainsi activés empruntent des voies de transduction spécifiques du type de récepteur, comme par exemple les voies PLCg1, RAS/MAPK, JAK/STAT ou des phosphatidyl-inositols.
1-2. Comment les cellules cancéreuses acquièrent-elles l'indépendance en signaux de croissance ?
L'indépendance en signaux de croissance, acquise par les cellules cancéreuses, peut provenir d'altérations moléculaires se situant à différents niveaux dans la cellule:
Au niveau des facteurs de croissance extracellulaires. Selon un mécanisme de stimulation autocrine, les cellules cancéreuses synthétisent les facteurs auxquels elles sont sensibles. Cette "boucle" de rétroaction positive est retrouvée entre autres dans les sarcomes et les carcinomes qui produisent du TGFa (Tumor Growth Factor a) (Kohler et al., 1992).
Au niveau des récepteurs membranaires qui ont une activité tyrosine kinase. Les récepteurs membranaires ainsi dérégulés induisent, aux cellules et de façon constitutive, des messages de survie et de prolifération. Ces voies de transduction seront détaillées dans la troisième partie de l'introduction.
Au niveau des molécules effectrices activées par les récepteurs membranaires. La dérégulation se situe en aval de l'activation du récepteur, au niveau des voies de transduction. La voie RAS/MAPK (Mitogen Activated Protein Kinases) est fréquemment touchée. 25% des tumeurs humaines présentent des protéines RAS ayant une structure altérée (Shannon 1995).
La prolifération cellulaire non contrôlée a été communément associée à une activité dérégulée des oncogènes puisque la transformation cellulaire est le phénotype observé dans les tumeurs. Cependant ce type de prolifération cellulaire doit être nécessaire mais pas suffisant pour maintenir à la cellule cancéreuse un phénotype cellulaire "transformé". En effet, de part la nature pléiotropique des récepteurs cellulaires et des protéines de signalisation qu'ils activent, la maintenance de la survie cellulaire et les voies impliquées dans la mitogenèse doivent jouer un rôle important dans la signalisation oncogénique (Evan et al., 2001). Dans certaines proliférations malignes la diminution de l'apoptose est un des événements importants qui déterminent une prolifération cellulaire incontrôlée. Les voies induisant l'apoptose ainsi que les protéines impliquées dans la survie cellulaire, sont donc fréquemment dérégulées dans de nombreux cancers (Reed 2001).
2-1. Les principales voies moléculaires dans l'induction de l'apoptose:
Les cellules peuvent mourir accidentellement, en raison d'une lésion aiguë, par désintégration ou nécrose. Par opposition, l'apoptose ou mort cellulaire programmée est un processus normal qui joue un rôle fondamental dans le développement et l'homéostasie cellulaire (Lockshin et al., 2001). Les cellules en apoptose subissent des modifications morphologiques caractéristiques: la cellule et son noyau rétrécissent, se condensent et se fragmentent en corps apoptotiques. La membrane plasmique ne se rompt pas mais exprime des signaux de mort reconnus par les macrophages, qui phagocytent les corps apoptotiques sans réponse inflammatoire (Zörnig et al., 2001).
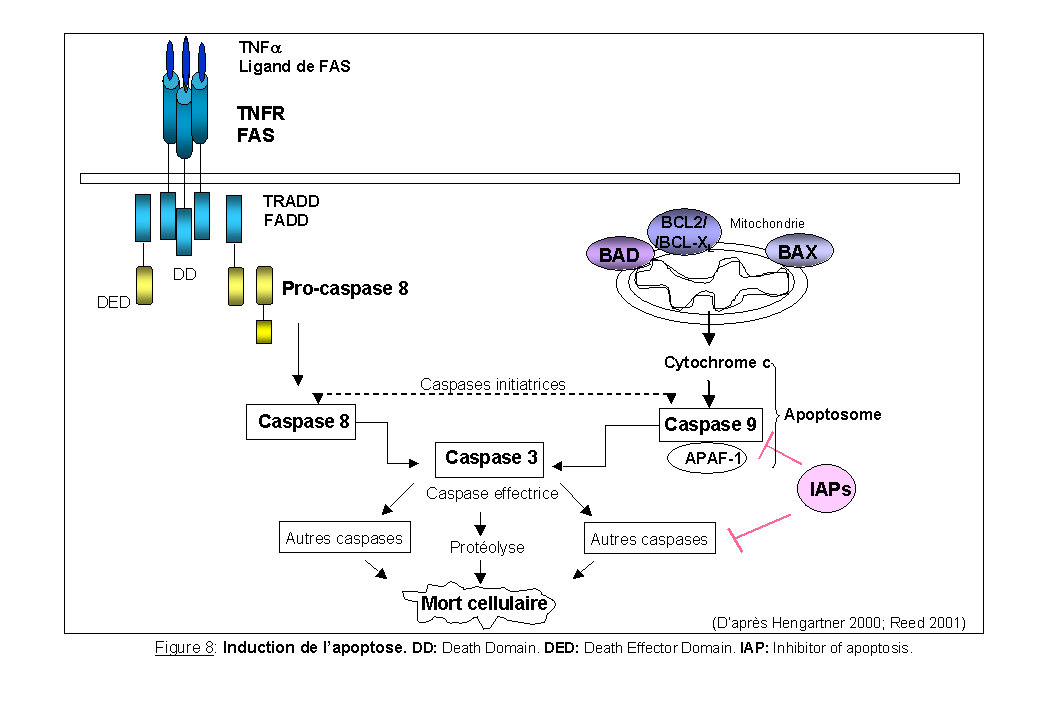
De nombreux signaux très différents, physiologiques ou pathologiques, intracellulaires ou extracellulaires, peuvent déclencher l'apoptose. Les dommages cellulaires, tels que les agents cytotoxiques, les radiations ionisantes et les chocs thermiques, induisent des signaux apoptotiques. L'apoptose peut être induite, par exemple, par la carence en facteurs de croissance et par certains récepteurs nucléaires (récepteurs aux glucocorticoïdes) activés par leur ligand. Deux voies principales peuvent être décrites dans l'induction de l'apoptose: l'activation des récepteurs membranaires de "mort" (comme TRAIL, les récepteurs de la famille TNF, TNFR1 ou le récepteur au ligand FAS nommé FAS/APO-1/CD95) et l'implication de la mitochondrie. Les signaux vont être intégrés par la cellule et de nombreuses voies de signalisation vont être ainsi activées aboutissant à l'activation des caspases (Figure 8).
Les caspases (pour Ced-3-like cysteine aspartate protease) de la famille ICE (interleukine-1b converting enzyme) sont des cystéines-protéases intracellulaires. Dix membres ont été identifiés (les caspases 2, 3, 6, 7, 8, 9, 10, 12, 13 et 14) qui fonctionnent comme des initiateurs et des effecteurs de l'apoptose (pour revue Cryns et al., 1998). Les caspases sont les effecteurs principaux de toutes les voies de transduction d'un signal apoptotique. Elle sont synthétisées sous forme de proenzymes inactifs. Les caspases 8 et 9 sont des "initiateurs" de l'apoptose (Engels et al., 2000).
Il est important de souligner que, contrairement à ce qui est présenté dans la Figure 8 très simplifiée, l'apoptose n'est pas le résultat d'une succession linéaire d'événements. De multiples voies de signalisation doivent communiquer et s'autoréguler.
2-1-1. L'apoptose induite par les "récepteurs de mort", TNFR1 et FAS/APO-1/CD95:
FAS appartient à la superfamille des récepteurs au TNF et possède un domaine cytoplasmique appelé DD (Death Domain), commun à TNFR1, qui est responsable de la transduction du signal (Figure 8). Après fixation du ligand, le récepteur se trimérise et entraîne une modification de conformation du domaine DD. Ce changement de conformation permet la fixation de la protéine TRADD (TNFR associated protein with DD) sur le TNFR et l'association de la protéine FADD (FAS associated protein with DD) avec FAS. Une fois fixé au récepteur, TRADD ou FADD s'associe à la pro-caspase 8 et l'active. La caspase 8 activée déclenche une cascade d'activation d'autres caspases dont la caspase 3 qui clivent alors certaines protéines cellulaires essentielles à la survie (Hengartner 2000).
2-1-2. L'apoptose induite par la mitochondrie:
Lorsque les cellules sont engagées en apoptose, une diminution du potentiel de membrane mitochondrial est observée et s'accompagne de l'ouverture de mégapores (Zörnig et al., 2001). Cette ouverture est responsable de la libération du cytochrome c dans le cytoplasme. Le cytochrome c, libéré par la mitochondrie, s'associe à la protéine régulatrice APAF-1, qui s'unit à la procaspase 9 pour former l'apoptosome (Hengartner 2000). Dans ce complexe, la caspase 9 est active et clive les protéases "effectrices" (les caspases 3, 6 et 7) qui entraînent l'activation de programmes conduisant à la mort cellulaire par apoptose (Zou et al., 1999). Une perte d'expression de la protéine APAF-1 est fréquemment retrouvée dans les mélanomes malins qui ont probablement acquis une résistance à l'apoptose (Soengas et al., 2001).
2-2. Les voies impliquées dans la survie cellulaire et leur surexpression dans certains cancers:
Les molécules qui induisent la survie cellulaire sont fréquemment surexprimées dans les cancers. La maintenance de la survie joue un rôle important dans la signalisation mitogénique. Dans les chimiothérapies, la résistance au traitement dépend de nombreux facteurs. Le recrutement de molécules antiapoptotiques est fréquent. Il augmente la capacité de survie des cellules, facilite la progression de la maladie et permet aux cellules cancéreuses d'échapper aux traitements induisant l'apoptose (Gorman et al., 2001). Les molécules antiapoptotiques contribuant au développement néoplasique et à la résistance aux chimiothérapies, incluent BCL2, PI3Kinase/AKT et RAS/MAPK (Makin et al., 2001).
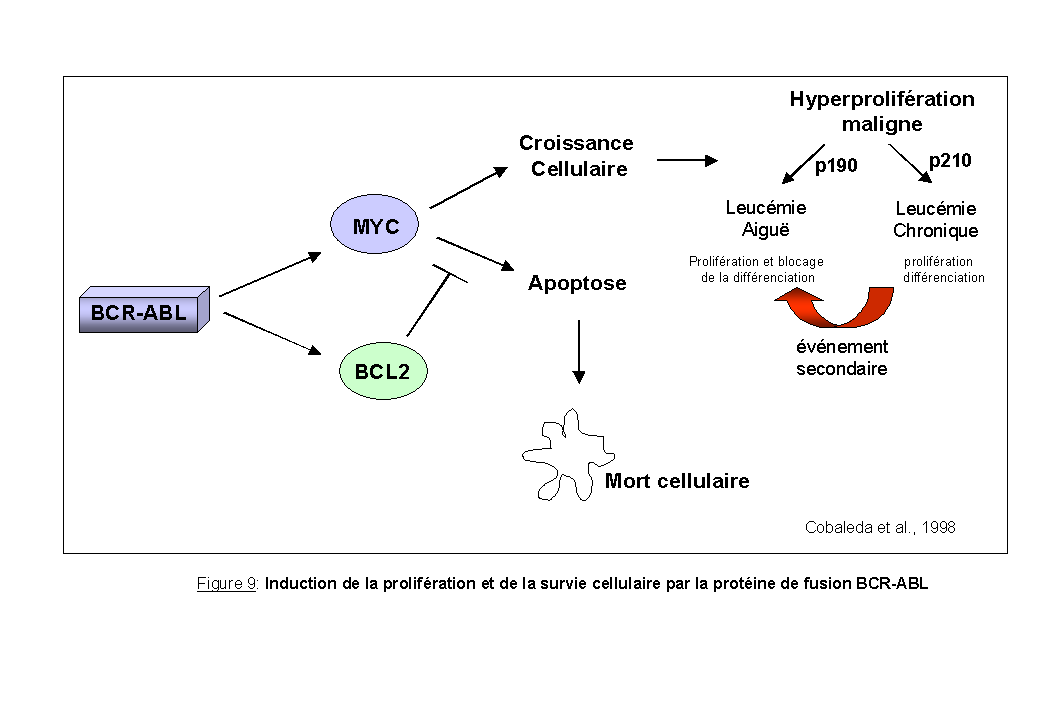
La protéine anti-apoptotique BCL2 est connue chez l'homme pour sa surexpression dans les lymphomes folliculaires avec une translocation t(14;18) (Tsujimoto et al., 1984). De fort niveaux d'expression de BCL2 ont été décrits dans une grande variété de cancers humains incluant des cancers de la prostate, du rein, du colon, de l'estomac, des neuroblastomes, des lymphomes non hodgkiniens et des leucémies chroniques et aiguës (Reed 1995). Dans le modèle de la leucémie myéloïde chronique, associée à la protéine de fusion BCR-ABL, l'induction de la survie cellulaire passe par une surexpression de la protéine BCL2 qui inhibe l'apoptose induite par c-MYC (Cobaleda et al., 1998) (Figure 9).
BCL2 appartient à une famille de protéines impliquées soit dans la survie (ce sont les molécules anti-apoptotiques) soit dans la mort cellulaire (ce sont les molécules pro-apoptotiques), (Pour revue Gross et al., 1999a) (Figure 10). La transfection de BCL2 dans des cellules myéloïdes ne donne aucun avantage sélectif de croissance mais supprime l'apoptose induite par la carence en facteurs de croissance.
Les molécules anti-apoptotiques de la famille BCL2 contiennent quatre domaines appelés BH (pour BCL2 Homology domain) (BH1 à BH4) correspondant à des hélices a et essentiels aux fonctions anti-apoptotiques de BCL2. Le domaine BH4, spécifique des molécules anti-apoptotiques, n'est pas retrouvé dans les molécules pro-apoptotiques. Les membres de la famille BCL2 ont la capacité de former des homodimères ou des hétérodimères et c'est le rapport entre le niveau des protéines pro- et anti-apoptotiques qui semble déterminer la sensibilité de la cellule à l'apoptose. BCL2 et certaines protéines de cette famille (BCL-XL par exemple) sont localisées sur les membranes externes de la mitochondrie, du noyau et du réticulum endoplasmique. Elles joueraient le rôle de canaux ioniques régulant ainsi le flux calcique et la libération du cytochrome c de la mitochondrie. BCL2 et BCL-XL exercent leur effet anti-apoptotique en partie en se liant à BAX, une molécule pro-apoptotique via leurs domaines BH1, BH2, BH3. Cette interaction empêche la libération du cytochrome c et l'activation du complexe caspase 9- APAF-1 (Figure 11).
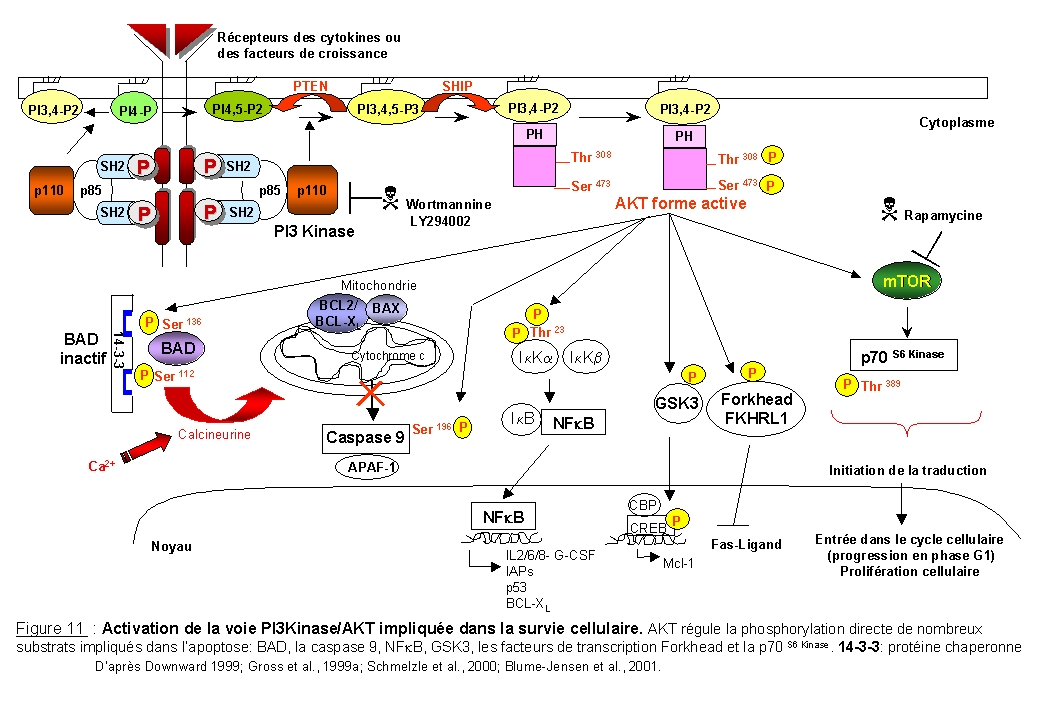
Les PI3Kinases appartiennent à une famille d'enzymes qui phosphorylent les lipides inositols des membranes en position 3 du groupe inositol (Wymann et Pirola 1998; Stein et al., 2000). Les PI3Kinases phosphorylent spécifiquement deux substrats: les phosphatidylinositol (4) phosphate (ou PI4P) et les phosphatidylinositol (4,5) bisphosphate (ou PIP2) générant ainsi des phosphatidylinositol (3,4) bisphosphate (PI3,4-P2) et des phosphatidylinositol (3,4,5) trisphosphate (ou PIP3), (Franke et al., 1997a), (Figure 11). Les phosphatidylinositols PI3,4-P2 et PI3,4,5-P3 sont absents à l'état normal dans les cellules quiescentes. Ils apparaissent suite à un stimulus extracellulaire et sont rapidement dégradés par des phosphatases (Franke et al., 1997a). PTEN est une phosphatase spécifique des phosphoinositides (elle génère des PIP2 à partir des PIP3) qui régule négativement l'activité des PI3Kinases. SHIP (SH2 containing inositol phosphatase) est une phosphatidylinositol 5' phosphatase qui permet la production de phosphatidylinositol (3,4) bisphosphate (PI3,4-P2) à partir de phosphatidylinositol (3,4,5) trisphosphate (PIP3).
Les PI3Kinases forment un complexe hétérodimérique composé d'une sous- unité catalytique de 110 kDa (p110a) et d'une sous- unité régulatrice/ adaptatrice de 85 kDa (p85a). La sous- unité p85 contient un domaine SH3, deux domaines SH2 et des motifs riches en résidus proline. La sous- unité p85 s'associe à des tyrosines phosphorylées du récepteur au niveau d'un motif consensus p-Y-X-X-M. Par son activité lipide kinase, la PI3Kinase entraîne la phosphorylation et l'activation de AKT/PKB, une sérine/thréonine kinase impliquée dans l'inhibition de l'apoptose (pour revues Fischer et al., 1998; Coffer et al., 1998; Datta et al., 1999; Brazil et al., 2001). Une inactivation de PTEN entraîne une phosphorylation constitutive de AKT (Cantley et al., 1999; Ramaswamy et al., 1999; Lu et al., 1999).
AKT a été initialement identifiée comme étant l'homologue cellulaire normal de l'oncogène v-akt transduit dans un rétrovirus murin. Cette sérine/thréonine kinase est exprimée de façon ubiquitaire et fait partie d'une famille de trois membres chez les mammifères: AKT1/PKBa, AKT2/PKBb, AKT3/PKBg. AKT est composée de trois régions distinctes: un domaine d'homologie à la pleckstrine (PH) en N-terminal suivi d'un domaine catalytique et d'une courte région régulatrice en C-terminal. Le domaine catalytique est apparenté à celui de la PKA et de la PKC.
Elle est activée dans beaucoup de types cellulaires par des facteurs de croissance (FGF2, PDGF, SCF, VEGF, NGF), des cytokines (IL-3, IL-2, IL-4, IL-5, IL-8) (Songyang et al., 1997) ainsi que par l'insuline (Burgering et al., 1995) et cette activation est bloquée par des inhibiteurs de la PI3Kinase, la wortmannine et le LY294002 (Walker et al., 2000). Les membres de la famille AKT sont surexprimés dans des tumeurs de l'ovaire (Yuan et al., 2000), du sein (Sun et al., 2001) ou du pancréas (Cheng et al., 1996) ce qui suggère que les kinases de type AKT pourraient participer à la résistance à l'apoptose fréquemment observée dans les cellules tumorales.
Comment est activé AKT ?
La PI3Kinase, activée par des facteurs de croissance ou de survie, génère des phospholipides (PI3,4-P2) insérés dans la membrane plasmique qui se fixent au domaine PH de AKT; ceci induit la translocation de AKT du cytoplasme vers la membrane plasmique (Figure 11), (Franke et al., 1997b). L'activation maximale de AKT se fait par la double phosphorylation des résidus thréonine Thr308 et sérine Ser473 par deux kinases différentes appelées PDK (phosphatidyl inositol dependent kinase). AKT sous forme activée phosphoryle de nombreux substrats cytoplasmiques qui ne sont pas encore, à l'heure actuelle, tous identifiés.
Les substrats de AKT:
AKT peut engendrer la survie cellulaire de trois façons différentes:
1. par la régulation des gènes de la famille BCL2 ou des caspases via des phosphorylations directes (phosphorylation de BAD, de la caspase 9).
2. par l'induction transcriptionnelle de gènes impliqués dans la survie cellulaire via l'activation de NFkB et l'inactivation de GSK-3.
3. par la répression transcriptionnelle de gènes pro-apoptotiques via la phosphorylation de facteurs de transcription séquestrés dans le cytoplasme.
1. Régulation des gènes de la famille BCL2 et des caspases:
La protéine BAD est une molécule pro-apoptotique formée d'un domaine BH3 (BCL2 Homology domain). Elle a été initialement identifiée par sa capacité à se lier à BCL2 et BCL-XL (un membre de la famille BCL2) (pour revues sur la famille BCL2 Gross et al., 1999a; Adams et al., 2001) (Figure 10). L'interaction de BCL2 ou BCL-XL avec BAD se fait au niveau du domaine BH3 de BAD (Kelekar et al., 1997). Cette interaction séquestre les pools de BCL2 et BCL-XL loin des protéines BAX, ce qui a pour conséquence le déclenchement de l'apoptose (Yang et al., 1995).
La protéine BAD, en présence de facteurs de survie, est phosphorylée sur trois résidus sérine 112, 136 et 155. AKT phosphoryle BAD sur sérine 136 de façon PI3Kinase dépendante. En effet, la phosphorylation de BAD est inhibée par des inhibiteurs spécifiques de la voie PI3Kinase (Datta et al., 1997; del Peso et al., 1997). BAD sous forme phosphorylée est alors séquestrée dans le cytoplasme par des molécules chaperonnes 14-3-3 (pour revue van Hemert et al., 2001). BAD ne peut plus interagir avec BCL-XL et n'exerce donc plus de fonction pro-apoptotique (Zha et al., 1996; Datta et al., 2000). La libération du cytochrome c est également inhibée (Figure 11).
La capacité qu'ont certaines cytokines à induire la survie cellulaire n'est cependant pas toujours corrélée avec l'activation de AKT et la phosphorylation de BAD. Trois lignées hématopoïétiques dépendantes de cytokines différentes, pour leur survie et leur prolifération, (Ba/F3/IL-3, FD-6/IL-4 et MC-9/GM-CSF) ont été utilisées pour étudier les relations existantes entre PI3Kinase, AKT, l'activité de BAD et la survie cellulaire. Dans les trois cas, les cytokines induisent une phosphorylation de AKT sensible aux inhibiteurs de la PI3Kinase. Cependant l'activation de AKT n'induit pas systématiquement la phosphorylation de BAD. Dans la lignée hématopoïétique FD-6 activée par l'IL-4 par exemple, la phosphorylation de BAD n'est pas détectée, laissant supposer qu'il existe d'autres voies de survie impliquées dépendantes et/ou indépendantes de la PI3Kinase (Scheid et al., 1998; Hinton et al., 1999).
Dans le but de comprendre le rôle des PI3Kinases dans des lignées hématopoïétiques Ba/F3 dépendante d'interleukine-3 (IL-3) pour leur survie et leur prolifération, Craddock et al., (1999) ont exprimé dans ces cellules une forme mutée de la PI3Kinase D p85 qui est dépourvue du site de fixation pour la sous- unité catalytique p110. Cette construction D p85 agit comme un dominant négatif bloquant l'activation catalytique de la sous- unité p110 (Hara et al., 1994). La conséquence majeure de l'expression de la D p85 est la réduction importante de la prolifération induite par l'IL-3, couplée à une diminution de la phosphorylation de AKT et de BAD. Ces effets ne sont pas associés à une augmentation de l'apoptose. Il doit donc exister des cibles autres que AKT et BAD pouvant engendrer un signal de survie dans ces cellules.
L'activation de la voie PI3Kinase/AKT entraîne la phosphorylation de la caspase 9. En effet, AKT est capable de phosphoryler sur sérine 196 une caspase 9 recombinante in vitro et d'inhiber son activité protéase en aval de la libération du cytochrome c (Cardone et al., 1998; Zhou et al., 2000). Les mécanismes exacts par lesquels AKT engendre la survie cellulaire ne sont pas encore bien définis. Contrairement aux résultats de Cardone et al., (1998) et de Zhou et al., (2000), une autre équipe a montré que AKT peut promouvoir un signal de survie indépendamment des caspases et de la phosphorylation de BAD en agissant sur la libération du cytochrome c (Kennedy et al., 1999). Il est possible que ces différents mécanismes agissent de façon alternative, comme des mécanismes de sauvegarde pour que AKT, in fine, puisse maintenir la survie cellulaire.
Toutes ces expériences montrent que l'effet de survie impliquant la PI3Kinase, AKT BAD et les caspases dépend des cytokines et du type cellulaire. Il est donc difficile de décrire une voie de transduction générale aboutissant à la survie cellulaire.
2. Induction transcriptionnelle de gènes impliqués dans la survie cellulaire:
NFkB est un facteur de transcription hétérodimérique qui est séquestré dans le cytoplasme par des protéines de la famille IkB (Karin 1999 pour revue). La phosphorylation de IkB permet son ubiquitination et sa dégradation par le protéasome. La dégradation de IkB libère NFkB qui est alors transloqué dans le noyau et qui active des gènes cibles: ce sont des gènes impliqués dans l'inflammation, dans les réponses au stress et dans l'inhibition de l'apoptose (Wang et al., 1996; Van Antwerp et al., 1996; Barkett et al., 1999 pour revue). Les gènes induits par NFkB pour promouvoir la survie ne sont pas encore clairement identifiés. Ils incluent un membre de la famille BCL2 (Bfl-1/A1) (Zong et al., 1999), des inhibiteurs de caspases (c-IAP-1 et c-IAP-2) et des facteurs TRAF1 et TRAF2 (TNFR-associated factor 1) (Chu et al., 1997; Wang CY et al., 1998).
AKT a été montré comme nécessaire et suffisant pour que le PDGF et le TNF induisent l'activité transcriptionnelle de NFkB (Romashkova et al., 1999; Ozes et al., 1999). AKT interagit directement avec IKKa (Kane et al., 1999) et phosphoryle cette kinase sur thréonine (Ozes et al., 1999) facilitant ainsi la dégradation des IkB. L'activation de NFkB n'est possible qu'en présence d'une protéine AKT ayant une activité kinase normale, un domaine PH intact et en présence d'une activité PI3Kinase endogène (Kane et al.,1999) (Figure 11).
La protéine GSK-3 (Glycogen synthase kinase-3) est une protéine sérine/thréonine kinase constitutivement active dans les cellules quiescentes et dont l'activité est inhibée par phosphorylation dans les cellules en prolifération. GSK-3 est impliquée dans divers processus biologiques incluant la synthèse du glycogène et l'activation de la b-caténine (pour revues Dominguez et al., 2001; Cohen et al., 2001). AKT phosphoryle et inhibe la GSK-3 en réponse à l'insuline (Cross et al., 1995) ce qui entraîne l'activation transcriptionnelle de gènes anti-apoptotiques tel que Mcl-1 (Figure 10 et Figure 10).
3. Répression transcriptionnelle de gènes pro-apoptotiques:
AKT phosphoryle un membre de la famille des facteurs de transcription Forkhead, FKHRL1/FOXO3a (Brunet et al., 1999). Selon la nomenclature des facteurs de transcription de la famille Forkhead, FKHRK1 appartient à la sous-famille FOXO (Forkhead box, group O). La conséquence majeure de cette phosphorylation est la modification de la localisation subcellulaire de FKHRL1. Sous forme phosphorylée, FKHRL1 est présent dans le cytoplasme et interagit avec des protéines chaperonnes 14-3-3. Cette interaction empêche l'entrée de FKHRL1 dans le noyau et l'activation des gènes pro-apoptotiques (comme le gène codant pour FAS-L par exemple).
mTOR et la voie PI3Kinase/ AKT:
L'effet transformant du couple PI3Kinase/AKT est relayé par la voie mTOR/p70 S6Kinase (Aoki et al., 2001). mTOR (mammalian Target of Rapamycin) appartient à la famille des protéines kinases PIKK (PIKinase-related kinase). Elle comprend dans sa partie C-terminale un domaine catalytique (phosphotransférase et lipide kinase) très similaire à celui de la PI3Kinase (p110). Cependant, il n'a été montré pour l'instant qu'une activité serine/thréonine kinase pour cette protéine (pour revues Thomas et al., 1997; Schmelzle et al., 2000). mTOR intervient dans une voie de signalisation mitogénique réglant l'initiation de la traduction. mTOR phosphoryle deux substrats majeurs impliqués dans l'initiation de la traduction protéique: 4E-BP (elF-4E binding protein) et p70 S6kinase:
Ø Le facteur d'initiation de la traduction 4E (elF4E) est, à l'état basal, séquestré et inhibé par la protéine 4E-BP1. mTOR en phosphorylant cette dernière, va permettre la libération de elF4E et l'initiation de la traduction. Les protéines ainsi synthétisées induisent directement ou indirectement la transition G1/S du cycle cellulaire (Brunn et al., 1997; Hara et al., 1997).
Ø La p70 S6kinase est une sérine/thréonine kinase qui est impliquée dans la régulation du cycle cellulaire, la synthèse protéique et la prolifération cellulaire (pour revue, Chou et al., 1995). Le substrat majeur de cette kinase est la protéine ribosomique S6. Sous forme phosphorylée, cette dernière initie la traduction des ARNm possédant un domaine riche en pyrimidine à leur extrémité 5'. Ces ARNm codent pour des protéines ribosomiques et des facteurs d'élongation (Jefferies et al., 1997). La p70 S6kinase est surexprimée dans des cancers du sein suggérant un rôle potentiellement oncogénique pour cette protéine (Barlund et al., 2000). Il a également été décrit une activation constitutive de la p70 S6kinase dans des cellules tumorales déficientes en PTEN (Lu et al., 1999).
Il existe un lien direct entre la voie PI3Kinase/AKT et mTOR. mTOR, par exemple, est phosphorylée constitutivement dans des lignées cellulaires de cancer de la prostate ayant des mutations inactivatrices dans PTEN ou surexprimant AKT3 (Sekulic et al., 2000). De plus, la croissance de ces lignées cellulaire est supprimée par la rapamycine, un inhibiteur spécifique de mTOR (Jefferies et al., 1997).
De nombreuses expériences ont été menées visant à montrer que la PI3kinase active la p70 S6kinase . Le blocage de l'activité de la PI3Kinase par la wortmaninne ou le LY294002 entraîne une inhibition de l'activation de la p70 S6kinase (Chung et al., 1994 ; Burgering et al., 1995). Une preuve complémentaire de l'activation de la p70 S6kinase par la PI3Kinase a été apportée par des travaux utilisant une forme constitutivement activée de la PI3Kinase. La surexpression d'une telle construction dans des expériences de transfection transitoire a montré une activation de la p70 S6kinase (Weng et al., 1995). Enfin, il a également été montré que l'activité de la p70 S6kinase est augmentée après activation de AKT (Burgering et al., 1995).
RAF-MAPK et la voie PI3Kinase/ AKT:
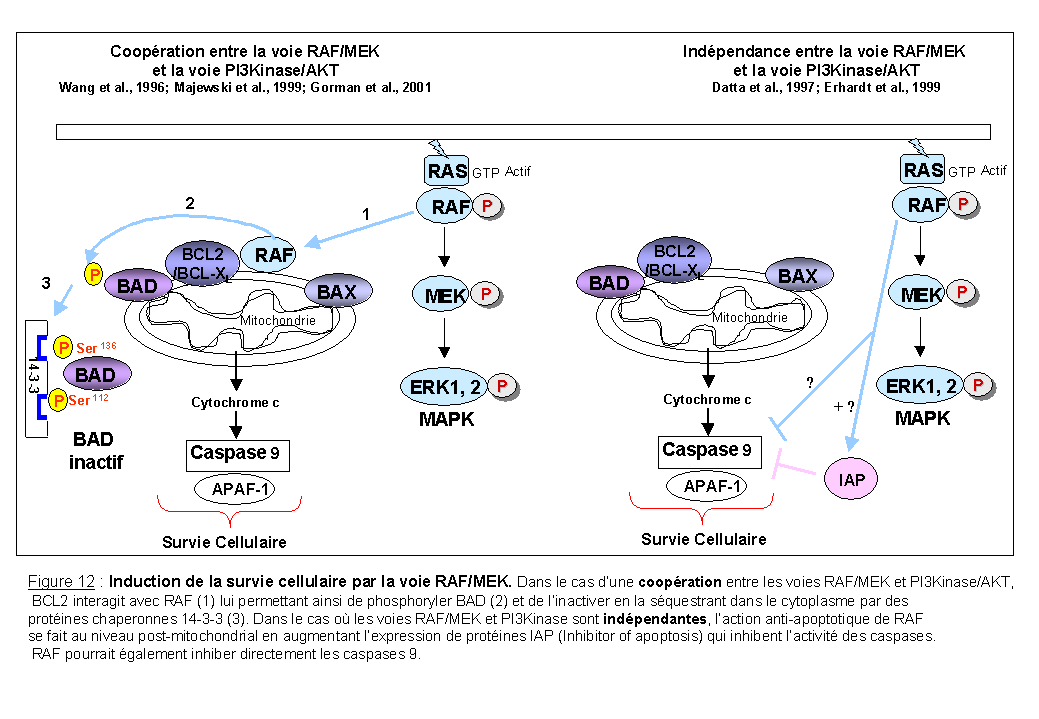
Une autre façon d'induire la survie cellulaire en inhibant la fonction pro-apoptotique de BAD est la voie des RAF-MEK (Figure 12 partie gauche). RAF1 supprime l'apoptose dans des lignées hématopoïétiques cultivées en absence de cytokine, probablement via une interaction avec BCL2. L'interaction de BCL2 avec RAF1, délocalise RAF1 aux membranes mitochondriales permettant ainsi à cette kinase de phosphoryler BAD (Wang H.G et al., 1996; Majewski et al., 1999). Von Gise et al., (2001) ont montré que cette activité de survie contrôlée par les MEK est dépendante de l'activité PI3Kinase dans les lignées hématopoïétiques. La phosphorylation de BAD par les MEK a été confirmée par l'utilisation d'un inhibiteur spécifique des MEK, le PD98059 (Pang et al., 1995) qui a pour conséquence l'inhibition de la phosphorylation de BAD dans des lignées dépendantes de l'IL-3 et du GM-CSF (Scheid et al., 1998). Von Gise et al., (2001) en utilisant un autre inhibiteur spécifique des MEK, le U0126, (Favata et al., 1998) montre que les signaux dépendant des MEK sont requis pour la croissance et la survie de cellules hématopoïétiques stimulées par l'IL-3. Ces résultats sont en contradiction avec ceux de Datta et al., (1997) qui montrent que l'inhibition des MEK n'a aucun effet sur la phosphorylation de BAD induite par le PDGF. Dans ce deuxième modèle (Figure 12 partie droite) l'action anti-apoptotique de la voie RAF/MEK/ERK se fait au niveau post-mitochondrial c'est-à-dire en aval de la libération du cytochrome c par la mitochondrie. Cette régulation post-mitochondriale de l'apoptose est retrouvée avec les protéines IAPs (Inhibitor of apoptosis) qui inhibent l'activité des caspases (Deveraux et al., 1998). Donc la voie RAF/MEK/ERK pourrait augmenter l'expression ou l'activité des protéines IAPs.
De nombreux facteurs contribuent au développement d'un cancer et dans la plus part des cas, la dérégulation de la prolifération cellulaire et la résistance à l'apoptose sont des événements importants qui déterminent la progression néoplasique (Evan et al., 2001).
Une meilleure compréhension des processus impliqués dans les cancers permet de développer des traitements. Ces traitements ciblent les protéines qui induisent des signaux de prolifération et les signaux de survie cellulaire (Kaufmann et al., 2000; Evan et al., 2001). Les voies impliquées dans la survie cellulaire, fréquemment surexprimées dans les cancers, ont été détaillées dans ce chapitre. Nous allons voir par la suite que les récepteurs membranaires à activité catalytique sont également la cible de dérégulation dans les cancers. Ces molécules, qui reçoivent des signaux de l'extérieur pour les transmettre à l'intérieur de la cellule et qui induisent de nombreux messages cellulaires comme la prolifération, la différenciation, la migration et l'apoptose, sont donc également des cibles thérapeutiques potentielles.
4. Références bibliographiques
Ce chapitre est issu de la THESE de Doctorat de Géraldine Guasch, 06 mars 2002, Université d'Aix-Marseille II
"Caractérisation moléculaire du syndrome myéloprolifératif 8p12 impliquant le gène FGFR1"
Directeur de Thèse : Mme le Docteur Marie-Josèphe Pébusque
© Copyright Université Aix-Marseille II 2002
Created: 29/04/2002
Last updated: Friday, 06-Feb-2026 15:23:25 CET
Author: Géraldine Guasch
Editors: Chantal Ginestoux and Marie-Paule Lefranc
IMGT Home page |
IMGT Repertoire (IG and TR) |
IMGT Repertoire (MH) |
IMGT Repertoire (RPI) |
IMGT Index |
IMGT Scientific chart |
IMGT Education |
IMGT Latest news ![]()



© Copyright 1995-2026 IMGT®, the international ImMunoGeneTics information system® | Terms of use | About us | Contact us | Citing IMGT
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}