
| IMGT Web resources |
|
| Here you are: IMGT Web resources > IMGT Education > Tutorials > Cancer |
Translocations chromosomiques et cancer
Géraldine GUASCH
U119 INSERM Laboratoire d'Oncologie Moléculaire
27 Bd Leï Roure 13009 Marseille
SOMMAIRE
1. Les anomalies chromosomiques dans les cancers
1-1. Les caractéristiques des cellules malignes
1-2. Les anomalies chromosomiques des cellules malignes
2. Les translocations chromosomiques
2-1. Les translocations chromosomiques dans les cancers en général
2-2. Les translocations chromosomiques dans les hémopathies malignes
2-2-1. Les hémopathies malignes
2-2-2. Les gènes fréquemment réarrangés dans les hémopathies malignes
2-2-3. Conséquences des translocations réciproques
3. Les protéines de fusion qui résultent des translocations chromosomiques
3-1. Les protéines de fusion impliquant les facteurs de transcription
3-1-1. Les protéines de fusion composées de domaines d’oligomérisation et d’un domaine d’activation transcriptionnel
3-1-2. Les protéines de fusion composées d’un domaine d’activation transcriptionnel fusionné à un domaine de liaison à l’ADN
3-1-3. Translocation impliquant les gènes codant pour les régulateurs de la transcription
3-2. Les protéines de fusion impliquant les protéines à activité kinase
3-2-1. Les protéines tyrosine kinase cytoplasmiques
3-2-2. Les récepteurs tyrosine kinase transmembranaire RTKs
4. Conclusions
5. Références bibliographiques
1. Les anomalies chromosomiques dans les cancers
La cancérogenèse chez l'homme est un processus "multi-étape" qui reflète la grande multiplicité des événements génétiques pouvant conduire à la maladie. Ces altérations génétiques induisent une transformation progressive d'une cellule normale en une cellule dérivée maligne. Les cellules cancéreuses ont des défauts dans leur programme de régulation qui gouverne la prolifération des cellules normales et l'homéostasie.
1-1. Les caractéristiques des cellules malignes
Le phénotype des cellules malignes est caractérisé par l'accumulation de trois éléments: l'immortalité, la tumorigénicité et l'instabilité génomique (Tlsty et al., 1992). L'instabilité du génome est un événement précoce important dans la progression tumorale: elle se manifeste par l'apparition de mutations ponctuelles et/ou de réarrangements de régions plus ou moins grandes du génome. Elle aboutit à la perte de la ségrégation fidèle des chromosomes aboutissant à la production de cellules aneuploïdes (perte ou gain d'un chromosome comme la trisomie 21 ou la monosomie X ) et hétéroploïdes (copies supplémentaires de tous les chromosomes comme la triploïdie 3n et la tétraploïdie 4n) (Lengauer et al., 1998; Ponder 2001).
1-2. Les anomalies chromosomiques des cellules malignes
Les cellules malignes présentent des anomalies acquises et parfois des anomalies constitutionnelles qui participent à la cancérogenèse. Les anomalies acquises sont présentes dans les cellules tumorales et absentes des tissus qui ne sont pas impliqués dans le phénotype malin. Ce sont des anomalies clonales apparues dans une seule cellule et présentes, en majorité, dans toutes les cellules de la tumeur considérée. Dans certains cas il peut y avoir perte du clone initial et donc perte des anomalies dans les cellules de la tumeur. La cellule portant l'anomalie présente des modifications génétiques lui conférant un avantage prolifératif.
Trois types d'anomalies chromosomiques sont retrouvées dans les cellules cancéreuses (Holliday 1989):
Ø le "bruit chromosomique", c'est-à-dire les anomalies chromosomiques dues au hasard, sans liaison ni conséquence directe avec le processus malin.
Ø les anomalies primaires, essentielles dans l'apparition des cellules tumorales.
Ø les anomalies secondaires, apparues au cours de l'évolution de la tumeur et importantes dans sa progression.
Les anomalies chromosomiques rencontrées dans les cancers peuvent être:
Ø des anomalies de nombre: perte et/ou gain d'un ou plusieurs chromosomes ce qui entraîne une modification de la quantité d'ADN totale de la cellule.
Ø des anomalies de structure: elles impliquent des cassures suivies de réarrangements aberrants. Elles peuvent être inter- ou intra-chromosomiques. On distingue les anomalies équilibrées (c'est-à-dire sans perte de matériel chromosomique), comme les translocations réciproques, les inversions et les anomalies déséquilibrées, comme les délétions, les duplications, les insertions et les isochromosomes (chromosomes dicentriques et en anneau). Les translocations réciproques associées à une délétion au point de cassure sont également des anomalies déséquilibrées.
Ces deux types d'aberrations chromosomiques peuvent coexister dans la même cellule cancéreuse et reflètent un caryotype complexe. Je ne détaillerai par la suite que les translocations réciproques car mon travail de thèse a porté plus particulièrement sur ce type d'anomalies équilibrées.
2. Les translocations chromosomiques
2-1. Les translocations chromosomiques dans les cancers en général
La découverte des translocations chromosomiques a transformé notre compréhension des mécanismes génétiques impliqués dans la leucémogenèse. Les translocations réciproques ont été identifiées dans les hémopathies malignes, les sarcomes et quelques tumeurs bénignes. Boveri en 1914 a été le premier à énoncer le concept suivant: un cancer dérive d'une seule cellule anormale. Il propose que des aberrations chromosomiques peuvent être la cause directe d'un cancer (pour revue Balmain 2001). Cette notion a été alors mis en évidence par la découverte du chromosome Philadelphie par Nowell et Hungerford en 1960 qui montrent pour la première fois qu'une anomalie chromosomique spécifique est liée à un type particulier de leucémie humaine, la leucémie myéloïde chronique. D'autres exemples confirment la notion que certaines anomalies chromosomiques sont spécifiques d'une tumeur maligne donnée. Dans les hémopathies malignes, la translocation t(15;17)(q21;q11-22) est associée à la leucémie aiguë promyélocytaire avec le gène de fusion PML-RARa (Kakizuka et al., 1991). Dans les tumeurs solides, la translocation t(11;22)(q24;q12) est retrouvée dans 85% des tumeurs d'Ewing et aboutit à la formation du gène de fusion EWS-FLI1 (Desmaze et al., 1997).
Dans de nombreux cancers, la présence de translocation spécifique est donc souvent importante pour le diagnostic et le pronostic et cette information a un impact direct sur la thérapie (Rowley 1999). De nouvelles translocations chromosomiques récurrentes sont sans cesse identifiées, il est donc difficile d'établir une liste exhaustive des aberrations chromosomiques dans les cancers. Un catalogue des aberrations chromosomiques mis en place par Mitelman et al., (2001) est disponible sous forme de database mais elle est encore incomplète. La découverte de ces anomalies chromosomiques conduit à l'identification de nouveaux gènes qui pourraient jouer un rôle dans la leucémogenèse.
Qu'est-ce qui favorise l'apparition des translocations ?
Les cassures peuvent être dues au hasard et la probabilité d'apparition est théoriquement la même dans n'importe quel point du génome. Des variations peuvent être causées par la structure chromatinienne (despiralisation, ADN-Z), par des sites spécifiques de coupure des DNA topoisomérase II, par des sites hypersensibles à la DNAse I ou par des séquences Alu répétées (Strissel et al., 1998).
Ces cassures sont associées à deux processus distincts: un processus de dérégulation de l'expression des gènes qui aboutit à la production d'une protéine normale en excès. Un processus de sélection qui génère des protéines de fusion anormales et qui ne favorise que les produits de fusion conférant un avantage prolifératif.
Les translocations équilibrées sont plus fréquemment étudiées dans les hémopathies malignes que dans les tumeurs solides. Les cellules hématopoïétiques malignes représentent un matériel de choix par leur facilité de culture. Au contraire, l'extension de l'analyse cytogénétique aux tumeurs solides s'est avérée beaucoup plus délicate en raison de la difficulté de leur mise en culture, de leur indice mitotique souvent faible, et fréquemment, du nombre et de la complexité de leurs anomalies chromosomiques. Les anomalies de structure équilibrées sont bien caractérisées dans les tumeurs d'origine mésenchymateuse (les sarcomes) et ont été plus rarement décrites dans les tumeurs d'origine épithéliale (les carcinomes) qui sont les plus retrouvées chez l'homme. Je ne décrirai par la suite que les translocations retrouvées dans les hémopathies malignes.
Après une brève description des hémopathies malignes, je décrirai les gènes fréquemment réarrangés dans ces tumeurs et les conséquences que peuvent avoir les translocations réciproques.
2-2. Les translocations chromosomiques dans les hémopathies malignes
2-2-1. Les hémopathies malignes
La dérégulation de l'homéostasie hématopoïétique peut être à l'origine des hémopathies malignes (pour revues Sawyers et al., 1991; Hanahan et al., 2000). Ce dysfonctionnement peut être consécutif à une expression inadaptée ou à des altérations structurales de certaines protéines suite à des mutations ponctuelles ou à des réarrangements chromosomiques. Les facteurs de croissance et leurs récepteurs, impliqués dans la régulation de l'hématopoïèse normale, sont fréquemment altérés dans les hémopathies malignes. La dérégulation de l'hématopoïèse normale peut être également due à une perte du contrôle de l'activité de facteurs participant à la régulation négative de la croissance normale des cellules hématopoïétiques.
Les leucémies humaines sont classées en fonction de leur degré de sévérité et du stade de maturation représenté. Quatre groupes peuvent être distingués:
Les leucémies aiguës résultent de la prolifération incontrôlée d'une population cellulaire clonale. Comme conséquence une amplification progressive du clone leucémique est observée, caractérisée par une dissociation complète entre la différenciation et la prolifération des cellules tumorales. Les cellules leucémiques (blastes) sont ainsi bloquées à différents stades de différenciation. Selon la classification FAB (French-American-British Cooperative Group) les leucémies aiguës sont de deux types: lymphoïdes (LAL) et myéloïdes (LAM) selon l'aspect morphologique et l'immunophénotype (Bennet et al., 1985 mis à jour par Flandrin 2001).
Les lymphomes (DeVita et al., 1999) sont des tumeurs dérivées du système lymphatique. Il existe plusieurs types de lymphomes, représentant la transformation de diverses classes de cellules lymphocytaires comme par exemple:
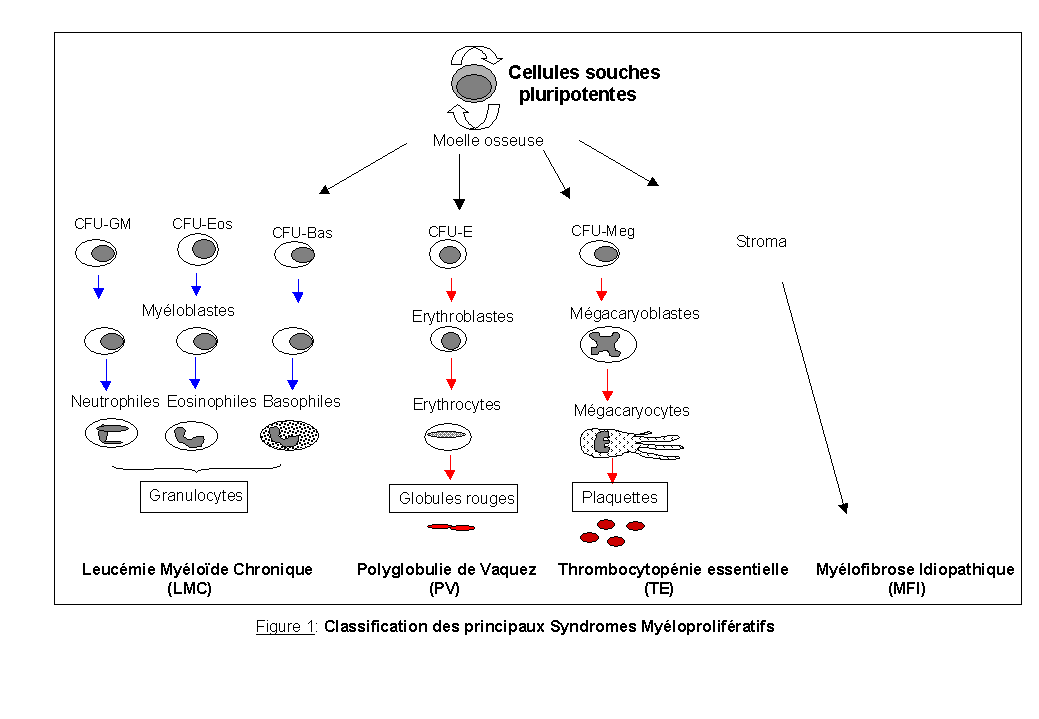
Les syndromes myéloprolifératifs (Dickstein et al., 1995; Albitar et al., 2000) sont des manifestations d'une anomalie clonale de la cellule souche hématopoïétique multipotente. Cette anomalie confère à la cellule souche un avantage prolifératif avec l'émergence d'un clone pathologique. Il se produirait dans un deuxième temps une lésion spécifique à chaque type de SMP, entraînant l'émergence de cellules progénitrices anormales. Contrairement aux leucémies aiguës, les SMP sont chroniques c'est-à-dire que les éléments myéloïdes gardent un potentiel de différenciation terminale normal ou quasi-normal. L'évolution des SMP se fait, après une période plus ou moins longue, vers la transformation en leucémie aiguë et/ou vers l'apparition de myélofibrose (fibrose médullaire sous forme réticulinique ou collagénique). Les SMP s'associent généralement à une reprise d'activité d'organes de l'hématopoïèse embryonnaire (rate et foie) (hépatosplénomégalie fréquemment associée).
Les SMP regroupent la leucémie myéloïde chronique (LMC), la polyglobulie de Vaquez (PV), la thrombocytopémie essentielle (TE) et la myélofibrose idiopathique (MFI) (Figure 1). Les cellules souches semblent être touchées à différentes étapes de leur différenciation par des facteurs dont l'origine reste inconnue, mais qui induirait une prolifération chronique maligne de la lignée granulocytaire (dans le cas de la LMC), de la lignée érythrocytaire (dans le cas de la PV) ou de la lignée plaquettaire (pour la TE).
La fibrose médullaire précoce ou myélofibrose avec métaplasie myéloïde, comme les autres SMP, est due à une transformation néoplasique de la cellule souche multipotente. Cependant, contrairement aux autres SMP chroniques, la caractéristique de cette affection est une myélofibrose qui étouffe l'hématopoïèse médullaire, entraînant une cytopénie périphérique et une importante hématopoïèse extra-médullaire dans la rate, le foie et les ganglions.
Récemment, de part leur caractère clonal, la leucémie chronique avec éosinophiles (Weide et al., 1997) et le syndrome hyperéosinophile idiopathique (Malbrain et al., 1996) sont considérés comme des formes de SMP. La leucémie neutrophile chronique est classée dans les SMP et il existe également des SMP atypiques inclassables.
Les syndromes myélodysplasiques sont le résultat d'anomalies qualitatives et quantitatives du fonctionnement médullaire se traduisant par des anomalies de maturation de la cellule souche et par des degrés divers d'hématopoïèse inefficace. Un trouble de la production des globules rouges, des polynucléaires et des plaquettes est observé, entraînant habituellement une cytopénie périphérique malgré une moelle hypercellulaire.
2-2-2. Les gènes fréquemment réarrangés dans les hémopathies malignes
Le clonage des points de cassure des translocations chromosomiques a permis l'identification de différentes classes d'oncogènes putatifs. Les oncogènes sont des gènes cellulaires appelés proto-oncogène devenus, suite à des modifications qualitatives ou quantitatives, des gènes "transformants" (Stehelin et al., 1976). Ce sont des gènes dont les mutations du type gain de fonction se manifestent à l'état hétérozygote. Ils peuvent être réarrangés par translocations réciproques ou par inversions. Ces oncogènes incluent des facteurs de croissance, des récepteurs aux facteurs de croissance, des protéines de la transduction du signal, des activateurs de la transcription et des protéines kinase. Les translocations ainsi créées activent ces gènes qui sont impliqués dans le contrôle de la prolifération et de la différenciation cellulaire.
2-2-3. Conséquences des translocations réciproques
Des études moléculaires des translocations ont permis de mettre en évidence deux mécanismes de leucémogenèse: la dérégulation de l'expression de gènes et la formation d'un gène de fusion. Chaque mécanisme, décrit ci-dessous, sera illustré par des exemples.
Dérégulation de l'expression de gènes:
Dans le cas de la dérégulation de l'expression de gènes, le point de cassure est situé le plus souvent en dehors de la partie codante du gène transloqué. Suite à la translocation, le gène est mis sous le contrôle des séquences régulatrices (enhancer et promoteur) d'un autre gène.
Ces translocations juxtaposent un promoteur fort, comme les loci d'immunoglobuline ou du TCR, à un gène régulateur de la différenciation ou de la survie cellulaire (Drexler et al., 1995; pour revue Willis et al., 2000). Elles conduisent à la surexpression d'un gène normal. Ces remaniements sont spécifiques des proliférations lymphoïdes malignes et en particulier les leucémies aiguës T (les gènes des immunoglobulines Ig ou des récepteurs des lymphocytes T y sont impliqués) et les lymphomes différenciés.
Les gènes affectés par ces translocations peuvent coder des facteurs de transcription comme MYC (Cleary 1991; Look 1997). Les prototypes de ces translocations sont la t(8;14), t(2;8) et t(8;22) du lymphome de Burkitt qui activent l'expression du gène MYC localisé en 8q24, sous le contrôle de différentes régions enhancer des immunoglobulines et aboutissent à la formation des fusions respectivement IgH-MYC, Igk-MYC et Igl-MYC (Haluska et al., 1987; Hecht et al., 2000). MYC est une protéine proto-oncogène à fonction de facteur de transcription. Il appartient à une famille de protéine contenant des motifs helix-loop-helix (HLH) qui permettent la fixation à l'ADN et une dimérisation avec des membres hétérologues de cette famille. Ces réarrangements engendrent la surexpression du gène MYC qui est transcriptionnellement silencieux à l'état normal.
Le gène BCL2, localisé en 18q21, peut être associé avec le gène des chaînes lourdes des immunoglobulines dans des lymphomes folliculaires ayant la t(14;18). BCL2 code pour une protéine mitochondriale impliquée dans la régulation de l'apoptose. Ce gène peut également être associé avec les gènes des chaînes légères des immunoglobulines Igk et l dans respectivement les translocations t(2;18) et t(18;22), présentes dans diverses proliférations lymphoïdes de la lignée B (Willis et al., 2000).
Les facteurs de croissance, impliqués dans la régulation de l'hématopoïèse normale, sont fréquemment altérés dans les hémopathies malignes. L'interleukine 3 (IL-3) par exemple est surexprimée dans un sous type de leucémies aiguës pré-lymphocytaires B avec éosinophilie suite à une translocation chromosomique t(5;14) (Meekers et al., 1990). Dans cette translocation, l'IL-3 est mis sous le contrôle des séquences régulatrices des chaînes lourdes des immunoglobulines.
Dans d'autres cas, l'allèle transloqué présente des mutations ponctuelles activatrices. C'est le cas du gène FGFR3 qui code pour un des quatre récepteurs à activité tyrosine kinase de la famille des facteurs de croissance du fibroblaste (FGF). FGFR3 est associé à la translocation t(4;14) dans le myélome multiple, une forme de prolifération lymphoïde chronique (Chesi et al., 1997). Cette translocation met le gène FGFR3 sous le contrôle de l'enhancer des IgH. Dans cette situation, l'hyperexpression et une mutation activatrice dans le domaine tyrosine kinase semblent être nécessaires pour déterminer l'initiation de la transformation maligne (Webster et al., 1997b; Chesi et al., 2001).
Formation d'un gène de fusion:
Les translocations aboutissant à la formation d'un gène de fusion, sont généralement retrouvées dans les leucémies myéloïdes et les leucémies lymphoïdes B. Deux gènes de fusion résultent de ces translocations équilibrées, ce qui conduit à l'expression d'une ou deux protéines chimères possédant des éléments de séquence des deux protéines impliquées (comme par exemple les protéines BCR-ABL et ABL-BCR résultant de la translocation t(9;22)). Le plus souvent, une seule des deux protéines de fusion a un rôle évident dans le processus leucémogène. Cette protéine de fusion présente un gain et/ou un effet dominant négatif sur la fonction des protéines parentales.
De nombreux gènes, incluant les gènes codant pour des régulateurs du cycle cellulaire, de la prolifération et de la différenciation cellulaire, sont impliqués dans le mécanisme de transformation maligne suite à des translocations réciproques. Parmi eux, deux grandes classes peuvent être décrites: les gènes codant pour des facteurs de transcription ayant des motifs de liaison à l'ADN et les gènes codant pour des protéines à activité tyrosine kinase. Les protéines de fusion qui résultent de ces translocations ont un rôle crucial dans la leucémogenèse.
3. Les protéines de fusion qui résultent des translocations chromosomiques
Les protéines de fusion créées suite aux translocations chromosomiques, peuvent agir différemment sur les cellules hématopoïétiques. Les facteurs de transcription chimères perturbent les voies normales de différenciation et les protéines tyrosine kinase activées confèrent un potentiel prolifératif et/ou anti-apoptotique aux cellules hématopoïétiques.
3-1. Les protéines de fusion impliquant les facteurs de transcription
Les facteurs de transcription, réarrangés dans les leucémies humaines, jouent un rôle important dans l'hématopoïèse normale. Les facteurs de transcription impliqués dans ces leucémies incluent CBF (Core Binding Factor)/AML, RARa (Retinoic Acid Receptor a), les membres de la famille HOX et les membres de la famille Ets (Voir pour revue Dash et al., 2001).
Les protéines de fusion impliquant les facteurs de transcription, peuvent être regroupées en deux classes suivant leur structure: celles formées d'un domaine d'oligomérisation et d'un domaine d'activation transcriptionnel et celles composées d'un domaine d'activation transcriptionnel fusionné à un domaine de liaison à l'ADN. Ces deux catégories de protéines de fusion seront décrites et illustrées par des exemples.
La conséquence de ces produits de fusion est la modification de la spécificité de liaison des facteurs de transcription. Le facteur de transcription chimérique agit comme un dominant négatif en inhibant la fonction du partenaire normal (Hiebert et al., 2001 pour revue). Ce cas de figure est illustré par les exemples suivants:
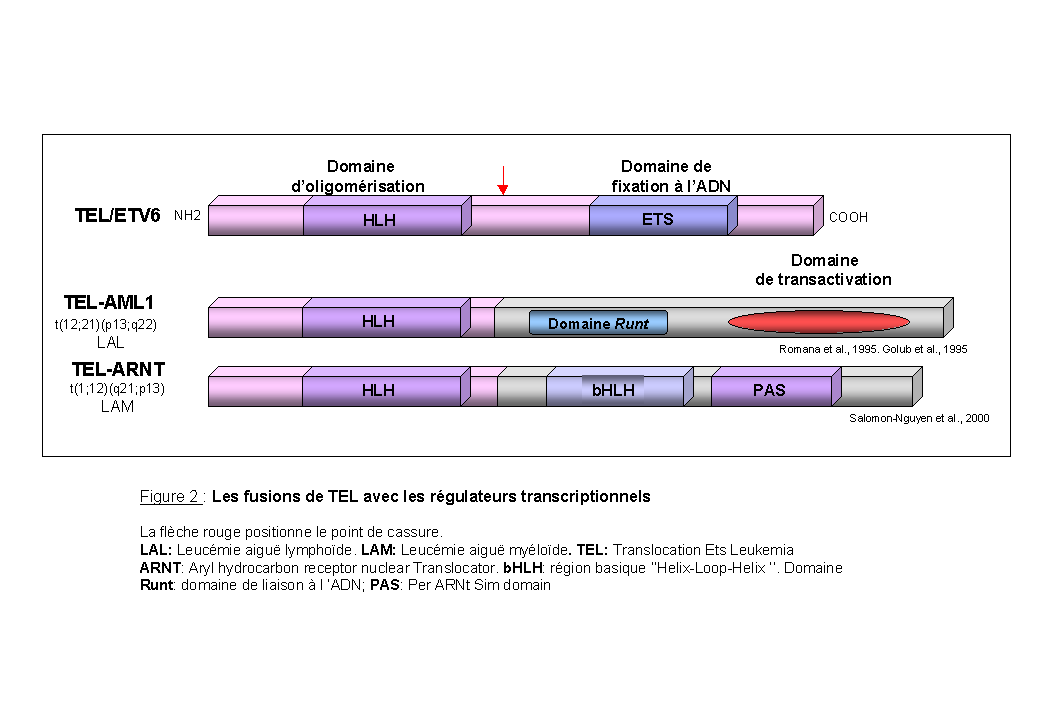
Les fusions de TEL avec les régulateurs transcriptionnels: (Figure 2)
Le gène TEL/ETV6 (Translocated ETS Leukemia) est un gène de la famille ETS qui code pour des régulateurs de la transcription. La protéine TEL est composée d'un domaine d'oligomérisation en N-terminal et d'un domaine ETS de liaison à l'ADN. Dans la translocation réciproque t(12;21)(p13;q22) associée à la leucémie aiguë lymphoblastique pré-B, et dans la translocation t(1;12)(q21;p13) associée à la leucémie aiguë myéloïde, deux protéines de fusion sont formées: respectivement TEL-AML1 et TEL-ARNT.
TEL-AML1 (Romana et al., 1995; Golub et al., 1995): le gène AML1 (CBFA2) code pour la sous-unité a du "Core Binding Factor" (CBF). Ce complexe a été initialement identifié comme un facteur de transcription liant une région régulatrice dans les "enhancers" de virus leucémogènes murins. Ces facteurs sont caractérisés par la conservation d'un domaine de 128 résidus, le domaine RUNT. Ce domaine est responsable de leur liaison spécifique à l'ADN, de leur localisation nucléaire et de leur interaction avec CBFb. Des expériences de transfection transitoire ont montré que AML1 augmente l'activité de l'enhancer du TCRb (Meyers et al., 1995). La protéine de fusion TEL-AML1 comporte les domaines d'oligomérisation de TEL et le domaine RUNT d'AML1. Hiebert et al., (1996) ont montré que la protéine de fusion TEL-AML1 inhibe l'activité d'un gène indicateur placé sous le contrôle de l'enhancer du TCRb et interfère avec l'activation de cet enhancer par AML1. Ainsi la protéine TEL-AML1 a un effet dominant négatif sur les propriétés normales d'AML1 et cette activité de répression est dépendante de l'intégrité du domaine HLH de TEL (Hiebert et al., 1996). Récemment, il a été montré que la protéine TEL-AML1 interagit avec le corépresseur SIN3A via le domaine HLH de TEL et via un domaine de 29 acides aminés en aval du domaine RUNT de AML1 (Fenrick et al., 1999).
TEL-ARNT (Salomon-Nguyen et al., 2000): le gène ARNT (Aryl hydrocarbon Receptor Nuclear Translocator) code pour un facteur de transcription de la famille bHLH-PAS impliqué dans la réponse à l'hypoxie et la réponse aux hydrocarbures aromatiques. La fusion TEL-ARNT a un domaine d'oligomérisation fonctionnel et la conséquence de cette fusion pourrait être de convertir ARNT en un répresseur transcriptionnel.
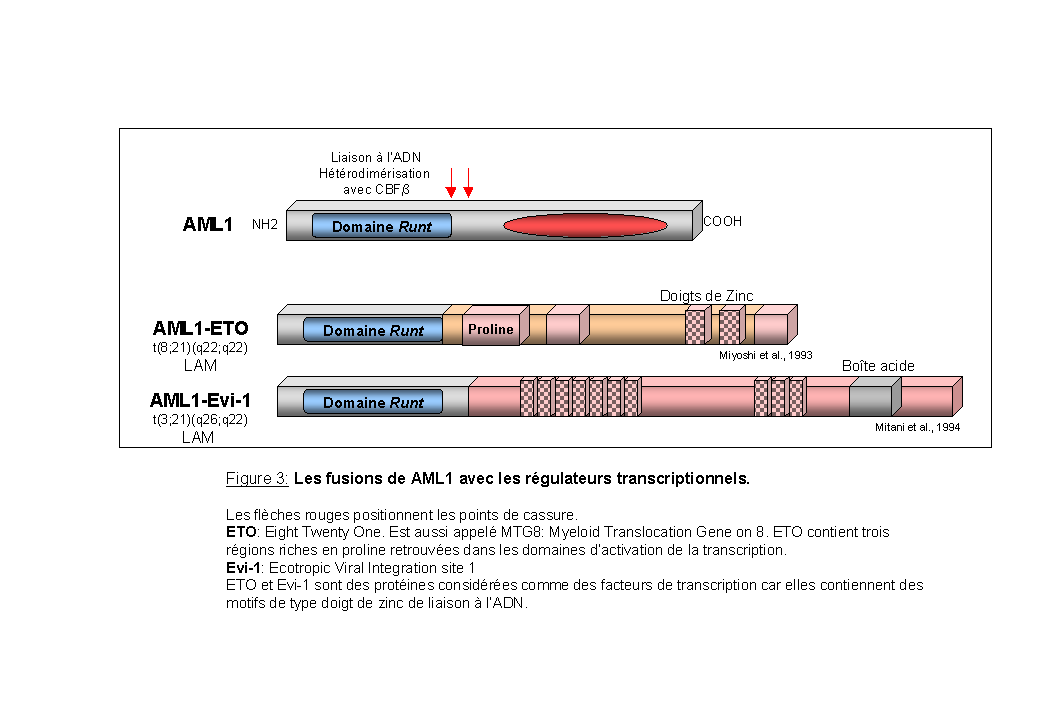
Les fusions de AML1 avec les régulateurs transcriptionnels: (Figure 3)
Dans les translocations réciproques t(8;21)(q22;q22) et t(3;21)(q26;q22) associées à une leucémie aiguë myéloïde, deux protéines de fusion sont formées, respectivement AML1-ET0 et AML1-EVI-1 (Miyoshi et al., 1993; Mitani et al., 1994), (Nucifora et al., 1995 pour revue). Les protéines de fusion conservent le domaine RUNT de AML1 fusionné à des domaines contenant des motifs en doigts de zinc. Ces protéines engendrent une accumulation nucléaire plus importante de la protéine CBFb que celle déterminée par la protéine AML1 sauvage (Tanaka et al., 1998). La répression de la fonction de la protéine AML1 sauvage par les protéines de fusion AML1-ETO et AML1-EVI-1, pourrait être un des mécanismes engendrant la leucémogenèse (Meyers et al., 1995; Tanaka et al., 1995).
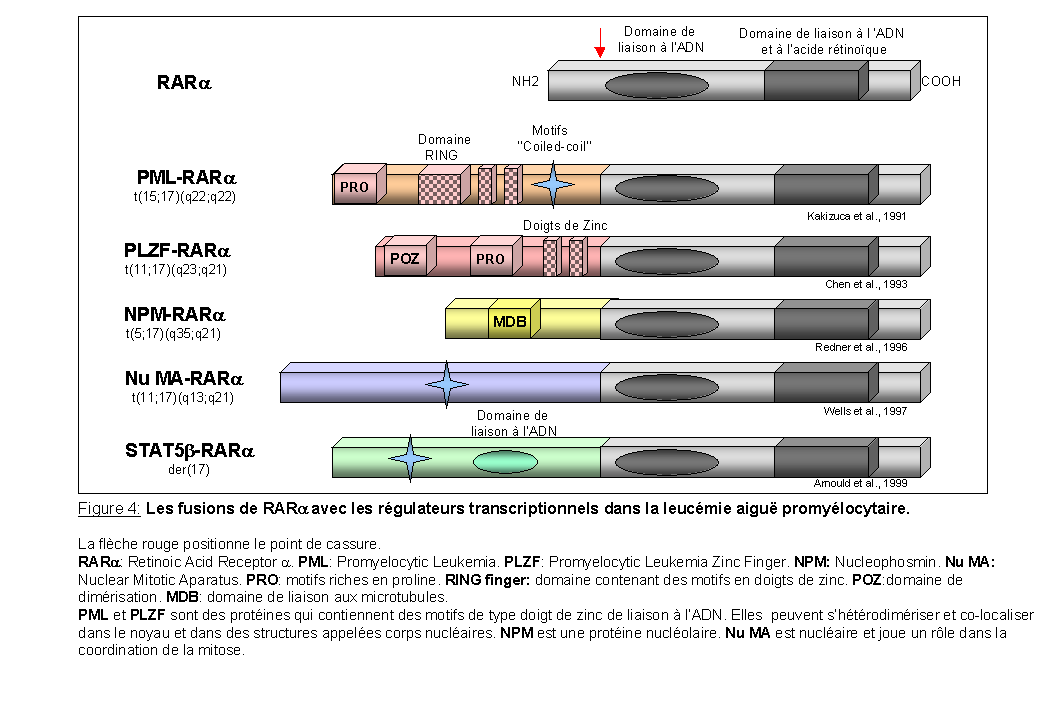
Les fusions de RARa avec les régulateurs transcriptionnels: (Figure 4)
La leucémie aiguë promyélocytaire est associée à différentes translocations chromosomiques impliquant toujours le gène codant pour le récepteur nucléaire à l'acide rétinoïque, RARa. Dans toutes les translocations décrites à ce jour, les protéines de fusion conservent le domaine de liaison à l'ADN et à l'acide rétinoïque de RARa. La translocation t(15;17), la plus fréquente, conduit à la formation de la protéine de fusion PML-RARa qui contient des motifs d'oligomérisation dans sa partie N-terminale (Kakizuca et al., 1991). Trois autres translocations, impliquant toujours RARa, ont été décrites chez de rares patients atteint de leucémie aiguë promyélocytaire: la translocation t(11;17) implique le gène PLZF qui code pour un répresseur transcriptionnel possédant des motifs de dimérisation (Chen et al., 1993), la translocation t(5;17) implique le gène NPM codant pour la nucléophosmine (Redner et al., 1996) et la translocation variante t(11;17) implique le gène NuMA dont la protéine est associée à la matrice nucléaire comme PML et NPM (Wells et al., 1997). Enfin un chromosome dérivatif der(17) implique STAT5b qui code pour un facteur de transcription activé par certaines cytokines (Arnould et al., 1999).
Les protéines de fusion X-RARa se comportent comme des répresseurs transcriptionnels en se liant avec une plus grande affinité que RARa aux co-répresseurs. Ceci a pour conséquence le blocage de la différenciation (He et al., 1998).
Des gènes codant pour des cofacteurs transcriptionnels sont souvent dérégulés dans les leucémies. Le gène PBX1, codant pour des protéines à homéodomaines, est réarrangé avec le facteur de transcription E2A dans la t(1;19) associée à une leucémie aiguë lymphoblastique pré-B (Kamps et al., 1990). Un autre exemple est la translocation t(7;11) associée à une leucémie aiguë myéloïde, qui fusionne le gène NUP98 (codant pour une nucléoporine, un composant du complexe du pore nucléaire qui permet le transport bi-directionnel des protéines et de l'ARN entre le noyau et le cytoplasme) à un gène HOXA9 codant pour une protéine à homéodomaine (Borrow et al., 1996b).
Les protéines de fusion résultantes E2A-PBX1 et NUP98-HOXA9, conservent le domaine d'activation transcriptionnel fusionné à un domaine de liaison à l'ADN. Ces deux protéines de fusion sont capables de transformer des fibroblastes NIH3T3 en culture (et de bloquer la diffenciation en immortalisant des myéloblastes dans le cas de E2A-PBX1) probablement en activant la transcription des gènes cibles de PBX1 et HOXA9 (respectivement Monica et al., 1994; Kamps et al., 1996 et Kasper et al., 1999). Dans le cas de E2A-PBX1, les propriétés transformantes de la protéine de fusion dépendent de la partie amino-terminale de E2A, responsable de l'activation de la transcription (Kamps et al., 1996).
3-1-3. Translocations impliquant les gènes codant pour les régulateurs de la transcription
Les régulateurs de la transcription, tels que CBP/p300 (Creb Binding Protein) et MLL (Mixed Lineage Leukaemia) sont également associés à des translocations dans les leucémies humaines, ce qui suggère un rôle critique joué par ces protéines dans la leucémogenèse.
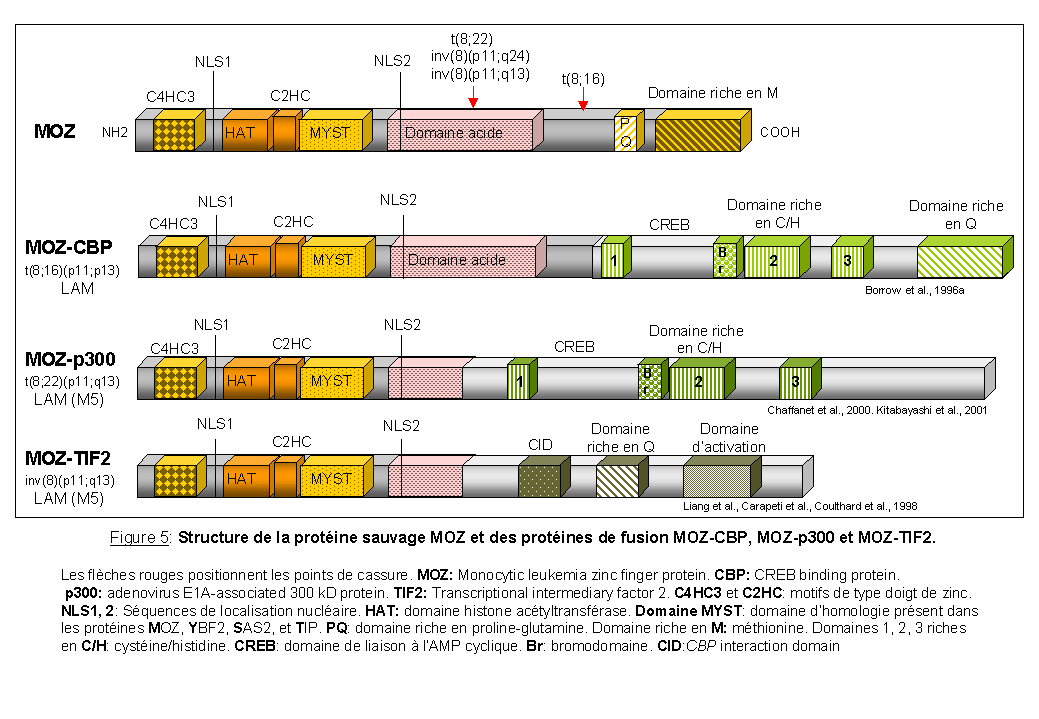
MOZ et les leucémies aiguës myéloïdes (Figure 5)
Le gène MOZ (Monocytic leukemia zinc finger protein) qui code pour une protéine ayant une activité acétyltransférase (Borrow et al., 1996a; Champagne et al., 2001), est réarrangé avec des gènes codant pour des coactivateurs transcriptionnels tels que CBP (CREB binding protein), p300 (adenovirus E1A-associated 300-kD protein) et TIF2 (Transcriptional intermediary factor 2). L'acétylation posttraductionnelle des histones est une des caractéristiques de la chromatine transcriptionnellement active. La protéine MOZ possède, entre autres, des motifs en doigt de zinc atypiques retrouvés dans des protéines de levures SAS2, YBF2/SAS3 (something about silencing) et dans la protéine humaine TIP (HIV TAT interactive protein) (Reifsnyder et al., 1996), un domaine histone acétyltransférase (HAT), un domaine acide et dans sa partie C-terminale un domaine riche en sérine-méthionine.
MOZ est réarrangé avec CBP dans la translocation t(8;16)(p11;p13) (Borrow et al., 1996a) et dans une translocation cryptique t(8;16) associée à une inversion péricentrique inv(8)(p11;q24) (Chaffanet et al., 1999). Dans la translocation t(8;22)(p11;q13) MOZ est réarrangé avec p300 (Chaffanet et al., 2000; Kitabayashi et al., 2001). Il est intéressant de noter que dans les translocations t(8;16) et t(8;22), les protéines de fusion conservent toute la partie N-terminale de MOZ contenant les motifs de type doigt de zinc et le domaine histone acétyltransférase en phase avec une grande partie de CBP et p300 qui présentent une structure très similaire (des domaines riches en cystéine- histidine et un domaine CREB de fixation à l'AMP cyclique) (Figure 5). CBP et p300 régulent la transcription par remodelage de la chromatine en acétylant les histones des nucléosomes. Une altération fonctionnelle de l'activité spécifique de ces deux protéines, suite aux translocations chromosomiques décrites, doit être un événement critique dans la leucémogenèse. La découverte des réarrangements des gènes MLL et CBP dans la translocation t(11;16)(q23;p13) (Taki et al., 1997) associée à des syndromes myélodysplasiques et des leucémies aiguës post-thérapeutiques, laisse supposer une importante participation du gène CBP, peut-être par un mécanisme "perte de fonction", dans la transformation maligne associée aux translocations t(8;16) et t(11;16).
Enfin, MOZ est réarrangé avec TIF2, un coactivateur des récepteurs nucléaires dans l'inversion péricentrique inv(8)(p11;q13) (Carapeti et al., 1998; Coulthard et al., 1998; Liang et al., 1998). La protéine de fusion MOZ-TIF2 (Figure 5) retient les domaines de fixation à l'ADN, les motifs de type doigt de zinc et le domaine HAT de MOZ liés aux domaines d'activation de TIF2 et aux domaines d'interaction avec CBP.
3-2. Les protéines de fusion impliquant les protéines à activité kinase
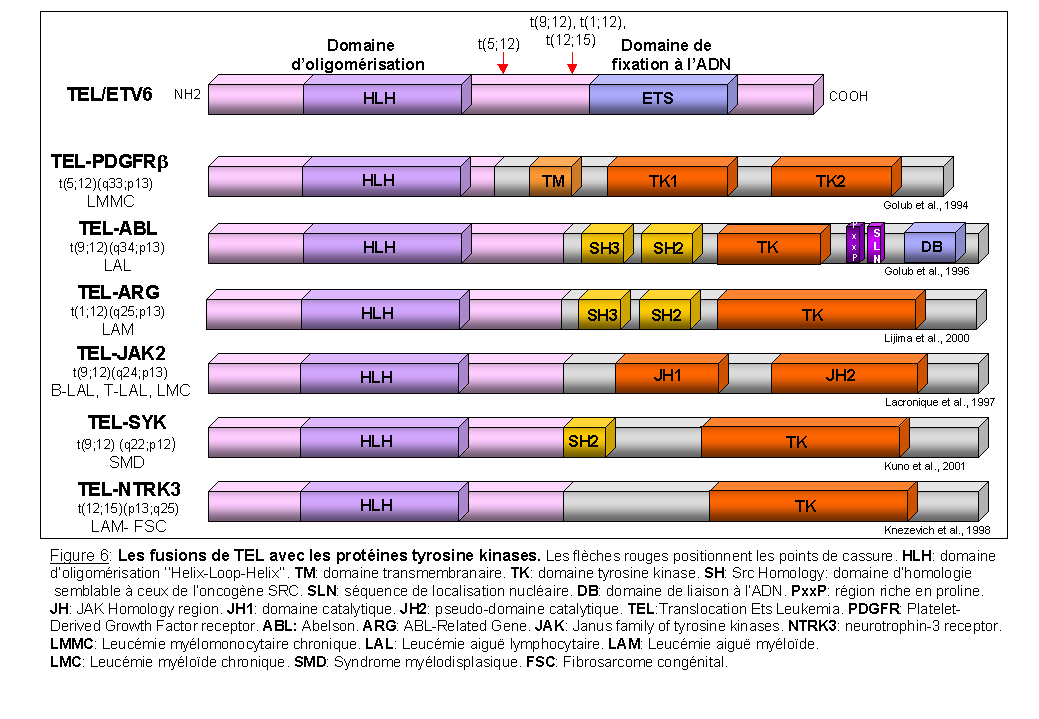
Dans les translocations impliquant les protéines à activité tyrosine kinase, la partie 5' du gène codant pour une protéine à activité tyrosine kinase, est remplacée par des séquences provenant d'un autre gène (Rodrigues et al., 1994). Les protéines de fusion sont composées de domaines d'oligomérisation et d'un domaine catalytique (Figures 6 et 7). La Figure 6 décrit cinq différentes protéines, retrouvées dans des hémopathies malignes, conservant le domaine d'oligomérisation en NH2 terminale de TEL/ETV6 fusionné au domaine catalytique de différentes protéines à activité tyrosine kinase. Dans ce cas, l'expression des gènes de fusion est sous le contrôle du promoteur de TEL, dont le profil d'activité et la régulation sont différents de ceux des gènes codant pour les protéines à activité tyrosine kinase.
Deux catégories de protéines tyrosine kinase peuvent être dérégulées suite à des translocations chromosomiques: les protéines cytoplasmiques et les récepteurs transmembranaires.
3-2-1. Les protéines tyrosine kinase cytoplasmiques:
ABL est une protéine tyrosine kinase composée d'une partie N-terminale possédant des domaines d'homologies SH semblables à ceux de l'oncogène SRC, impliquée dans la transmission du signal, et une partie C-terminale de liaison à l'ADN (pour revue Smith et al., 2002). Dans la leucémie aiguë lymphoblastique avec la translocation t(9;12)(q34;p13) ABL est fusionné au domaine HLH de TEL (Figure 6). Dans diverses leucémies avec la translocation t(9;22), ABL est fusionné au domaine d'oligomérisation de BCR (Figure 7). En fonction de l'endroit où se produit le point de cassure sur le gène BCR trois gènes de fusion sont crées (Figure 7). Le gène de fusion qui code pour une protéine BCR-ABL de 190 kDa est retrouvé en majorité dans la leucémie aiguë lymphoblastique (Melo 1996). Il est également associé dans très peu de cas, à des leucémies myéloïdes chroniques atypiques avec monocytose (Melo et al., 1994), des leucémies aiguës myéloïdes (Kurzrock et al., 1987) et des lymphomes (Mitani et al., 1990). Le deuxième gène de fusion crée code pour une protéine BCR-ABL plus grande, de 210 kDa, caractéristique de la leucémie myéloïde chronique. Cette protéine de 210 kDa est également retrouvée dans des leucémies aiguës lymphoblastiques (Deininger et al., 2000), plus rarement dans des leucémies aiguës myéloïdes (Kantarjian et al., 1991) et des myélomes (Martiat et al., 1990). La protéine p210 BCR-ABL contient en plus un domaine important DBL-like d'échange GDP/GTP (domaine d'homologie à la protéine DBL) qui pourrait expliquer les différences cliniques observées entre les différentes protéines BCR-ABL (Deininger et al., 2000; Maru 2001). Enfin une troisième forme BCR-ABL de 230 kDa a été observée en majorité dans les leucémies chroniques à polynucléaires neutrophiles (Pane et al., 1996) et dans quelques cas de leucémie myéloïde chronique (Wilson et al., 1997). La protéine p230 BCR-ABL contient pratiquement toute la protéine BCR avec les deux tiers du domaine C-terminal GAP à activité GTPase. Cette différence de structure pourrait expliquer la maturation granulocytaire normale dans ces formes particulières de leucémie myéloïde chronique.
La caractéristique commune entre les protéines de fusion TEL-ABL et BCR-ABL est la présence d'un domaine d'oligomérisation en N-terminal fusionné au domaine tyrosine kinase de ABL. Le domaine d'oligomérisation est crucial pour les propriétés transformantes des protéines de fusion (McWhirter et al., 1993; Golub et al., 1996). En effet, suite à leur oligomérisation, il y a activation constitutive du domaine kinase de ABL et stimulation des voies de transduction similaires (Okuda et al., 1996; Voss et al., 2000).
Deux autres exemples de protéine tyrosine kinase cytoplasmiques dérégulées suite à des translocations chromosomiques peuvent être décrites: la protéine JAK2, associée aux récepteurs des cytokines, dérégulée suite à la translocation t(9;12)(q24;p13) (Lacronique et al., 1997) et la protéine SYK, associée à de nombreux récepteurs dans les cellules hématopoïétiques, réarrangé dans la t(9;12)(q22;p12) (Kuno et al., 2001). Dans ces deux translocations, TEL est le gène partenaire. Suite à l'oligomérisation déterminée par le domaine HLH de TEL, il y a activation constitutive du domaine tyrosine kinase dans les protéines de fusion responsable de la transformation maligne.
3-2-2. Les récepteurs tyrosine kinase transmembranaire RTKs:
Les gènes codant pour des récepteurs à activité tyrosine kinase sont fréquemment réarrangés suite à des translocations réciproques (Schlessinger et al., 1992). C'est le cas du récepteur au PDGF (Platelet derived Growth Factor) PDGFRb dans la leucémie chronique myélomonocytaire avec la translocation t(5;12) (Golub et al., 1994), du récepteur à la neurotrophine 3, NTRK3 (ou TRKC) dans le fibrosarcome congénital et la leucémie aiguë myéloïde associé à la translocation t(12;15) (Knezevich et al., 1998) (Figure 6), et du récepteur ALK (anaplastic lymphoma kinase), dérégulé suite à la translocation t(2;5) associé à un lymphome non-hodgkinien (Morris et al., 1994; Hernandez et al., 1999).
Dans les protéines de fusion ainsi créées, il y a perte des séquences codant pour les domaines extracellulaire et transmembranaire des RTKs, domaines responsables respectivement de la fixation du ligand et de leur localisation au niveau de la membrane, et il y a conservation des séquences codant pour le domaine catalytique. Les nouvelles séquences juxtaposées en 5' favorisent la dimérisation, comme dans les fusions TEL-PDGFRb et TEL-NTRK3 où la partie du gène TEL code pour une région "Helix-Loop-Helix" (HLH) (Figure 6). Ceci est également retrouvé dans les fusions NPM-ALK et TFG-ALK où le domaine NH2 terminal contenant des hélices hydrophobes ou motifs "Coiled-Coil" est responsable de la dimérisation et de l'activation constitutive de la kinase (respectivement Bai et al., 1998; Hernandez et al., 1999). Les conséquences de ces réarrangements sont importantes pour la fonction du RTK (Blume-Jensen et al., 2001). En effet, l'activation du récepteur devient constitutive et donc non spécifique, non régulée, et indépendante de la fixation du ligand. Les séquences de dimérisation engendre une activation constitutive du domaine tyrosine kinase. La perte du domaine transmembranaire délocalise les protéines de fusion vers d'autres compartiments cellulaires. Une conséquence pourrait être que ces protéines de fusion, en plus d'activer constitutivement les voies de signalisation situées normalement en aval du récepteur sauvage, recruteraient et/ou induiraient la phosphorylation de substrats spécifiques, distincts de ceux normalement régulés par le récepteur membranaire sauvage. De plus, dans les protéines de fusion, la kinase est sous le contrôle d'un promoteur différent ce qui peut perturber l'expression normale du RTK.
Je développerai plus en détail les RTKs appartenant à la famille des récepteurs des facteurs de croissance du fibroblaste (FGFR) dans la dernière partie de l'introduction.
Les hémopathies malignes peuvent être associées à des altérations moléculaires. Les plus fréquentes sont les translocations chromosomiques aboutissant à la dérégulation de l'expression d'un gène normal ou à la synthèse d'une protéine de fusion oncogénique possédant des éléments de séquences de deux protéines normales.
Les produits normaux des gènes impliqués dans les réarrangements chromosomiques interviennent dans de nombreux processus cellulaires (pour revue Rabbitts 2001):
Cependant les voies d'activation ne sont pas variées:
Selon le type de maladie, les gènes impliqués dans les translocations chromosomiques et les voies de transduction activées par les protéines de fusion résultantes sont remarquablement communs. Dans le cas d'une leucémie chronique les gènes impliqués seront plutôt des gènes intervenant dans la signalisation cellulaire et les mécanismes mis en jeu seront la survie cellulaire et la résistance à l'apoptose. Dans le cas d'une leucémie aiguë, les gènes cibles seront impliqués dans le contrôle de la différenciation et de la prolifération cellulaire avec in fine un blocage de la différenciation et un mécanisme de prolifération.
5. Références bibliographiques
Ce chapitre est issu de la THESE de Doctorat de Géraldine Guasch, 06 mars 2002, Université d'Aix-Marseille II
"Caractérisation moléculaire du syndrome myéloprolifératif 8p12 impliquant le gène FGFR1"
Directeur de Thèse : Mme le Docteur Marie-Josèphe Pébusque
© Copyright Université Aix-Marseille II 2002
Created: 29/04/2002
Last updated: Friday, 06-Feb-2026 15:23:25 CET
Author: Géraldine Guasch
Editors: Chantal Ginestoux and Marie-Paule Lefranc
IMGT Home page |
IMGT Repertoire (IG and TR) |
IMGT Repertoire (MH) |
IMGT Repertoire (RPI) |
IMGT Index |
IMGT Scientific chart |
IMGT Education |
IMGT Latest news ![]()



© Copyright 1995-2026 IMGT®, the international ImMunoGeneTics information system® | Terms of use | About us | Contact us | Citing IMGT
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}