Introduction
IMGT/PhyloGene [2] is part of
IMGT, the international ImMunoGeneTics information system®
https://www.imgt.org [3].
IMGT/PhyloGene, created by LIGM
(Université Montpellier II, CNRS) is an IMGT tool for sequence analysis, on the Web since 10 February 2002.
IMGT/PhyloGene is an easy way to use tool for phylogenetic analysis of IMGT standardized reference sequences.
IMGT/PhyloGene is an on line software package which computes and draws phylogenetic trees for immunoglobulin (IG) and T-cell receptor (TR) V-REGION nucleotide sequences.
IMGT/PhyloGene consists of 5 complementary tools:
- A selection tool which allows to interactively and progressively select the IG and TR sequences to analyse.
- An evolutionary distance calculation tool which computes a distance matrix [4] from the selected sequences.
- A tree building tool which uses the Neighbor-Joining algorithm [5] to build a phylogenetic tree from the previously calculated distance matrix.
- A tree drawing tool which provides a graphical representation of a rooted phylogenetic tree.
- A label subsequences viewing tool which displays the subsequences corresponding to V-REGION related labels at the tips of a phylogenetic tree.
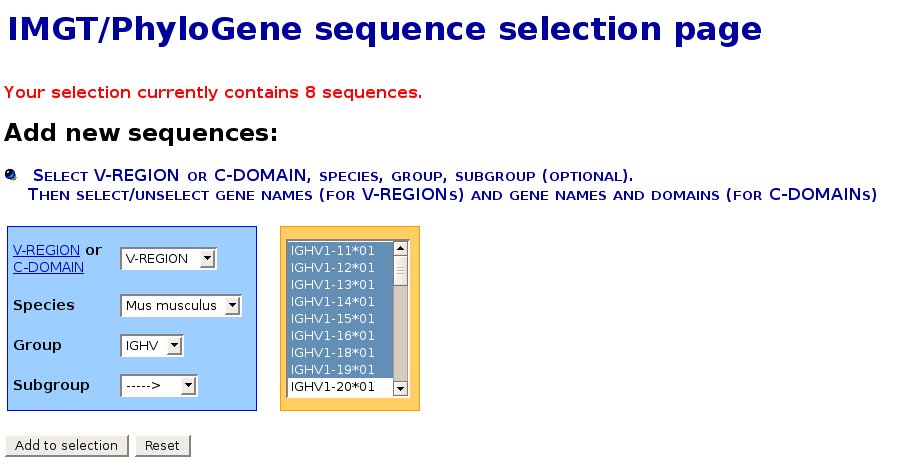
IMGT/PhyloGene sequence selection page
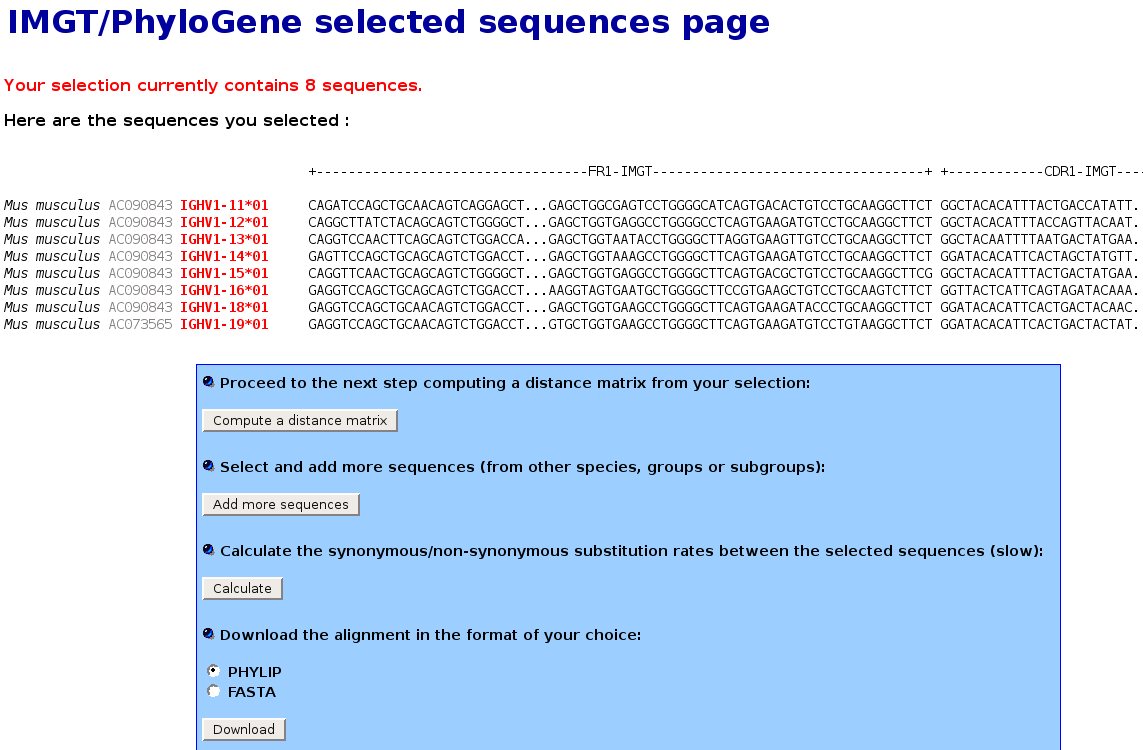
IMGT/PhyloGene selected sequences page
- This page shows the selected sequences. The IMGT unique numbering allows to get an automatic alignement of the sequences. The delimitations of the FR-IMGT and CDR-IMGT are also shown.
Note that the CDR-IMGT will be removed for the phylogenetic analysis by default. Indeed, the various lengths do not allow a meaningful alignment. Since the IMGT numbering provides clear information about the position and length of these regions, it is very easy to omit them.
- In the following screenshot, genes from the from the mouse IGHV1 subgroup
have been selected :
- At the bottom of this page,
- the "Add more sequences" button allows to select and add more sequences from other species, groups or
subgroups. Note that this procedure can be repeated this as many times as needed.
- the "Calculate" button allows to calculate the synonymous non synonymous substitution rates between
sequences (slow). At the top of the IMGT/PhyloGene substitution rates result page are shown the average
values for the Ks and Ka of all the analysed sequences. The Ks and Ka between the sequences, compared two by
two, are displayed in the table below.
- the "Download" button allows to download the alignment in the format of your choice (PHYLIP or FASTA).
- When all the sequences to analyse have been included into the selection, press the "Compute a distance matrix" button to proceed to the next step.
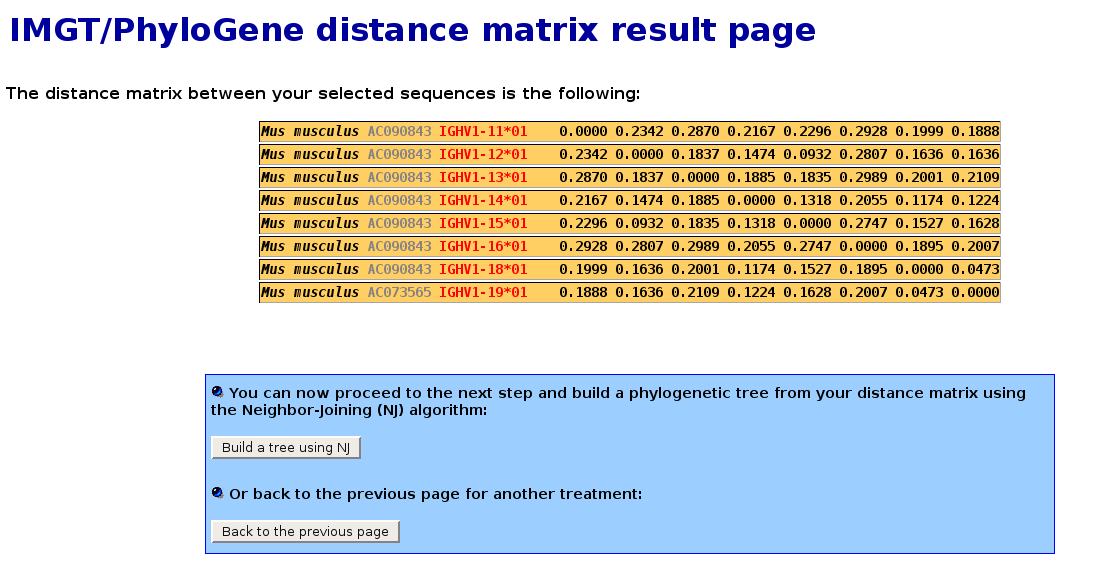
IMGT/PhyloGene distance matrix result page
- This page is only meant to show the distance matrix that has been calculated from the selected sequences.
- Distance between DNA sequences belonging to the selection are calculated using the Kimura 2-parameters
model of evolution [4]. Although rather simple,
this model of evolution is very often used in phylogenetic analysis. For example, it comes as a default option in the DNADIST program
from the PHYLIP package [6].
-
In the following screenshot, the distance matrix between genes from the mouse IGHV1 subgroups is shown :
- Pressing the "Build a tree using NJ" button will proceed to the next step and build a phylogenetic tree from this distance matrix.
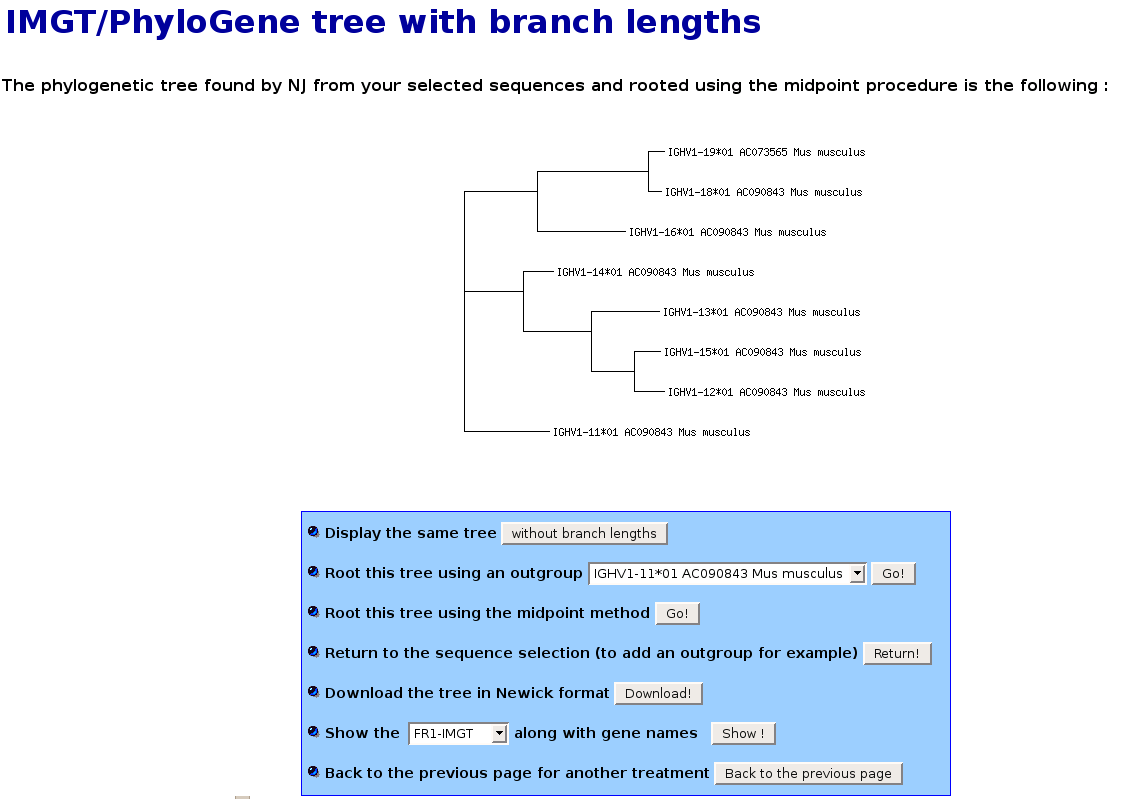
IMGT/PhyloGene tree drawing page
-
This page shows the phylogenetic tree that has been calculated using the Neighbor-Joining algorithm [5]. The NJ algorithm is one of the most famous algorithm
for reconstructing phylogenetic trees. Besides giving good results in term of reconstruction, it is very fast, and therefore very well suited to
online phylogenetic analysis, where nearly immediate response times are needed. Interested readers should
refer to the literature ([5] [7] [8] for example)
for further details about
the NJ algorithm.
-
Once constructed, the NJ tree is rooted using the midpoint procedure, which consists in locating the root halfway in the tree from the most divergent
sequences. Another way of rooting a phylogenetic tree is to include an outgroup in the form of one or several sequences whose duplication or speciation
is anterior to every other duplication or speciation that has occurred among the rest of the selection. This can be done easily with the selection page
described above (by pasting that (or those) sequence(s) in the text area, for example).
Once rooted, the tree is then drawn in the form of a horizontal phenogram. This kind of representation, very common in the literature, allows for easy visualisation of sequence groupings within the tree.
-
The default tree representation, IMGT/PhyloGene "tree with branch lengths", incorporates branch lengths : this means that the branch lengths of the represented tree have a meaning in term of evolutionary distance.
-
In the following screenshot, the NJ tree of the mouse IGHV1 subgroups is represented with branch lengths :
- At the bottom of this page, a button allows you to draw the same "tree without branch lengths" : in that kind of representation,
branch lengths are artificially arranged so as to align gene names and have no meaning in term of evolutionary distance.
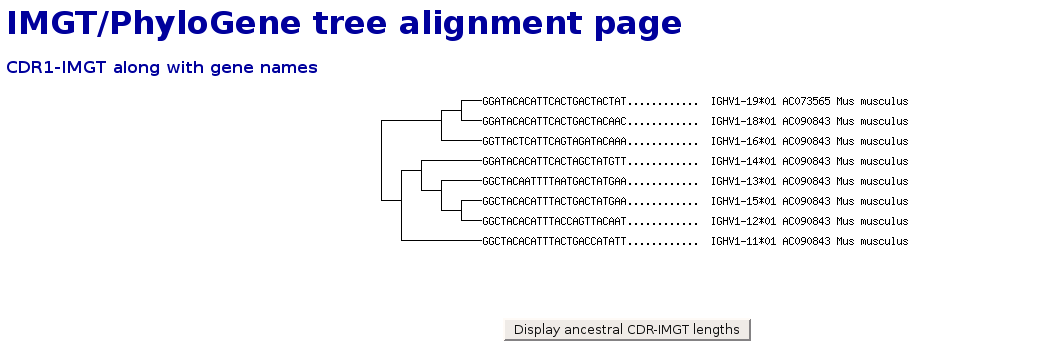
IMGT/PhyloGene tree alignment page
- In addition to the tree, it is possible to view, along with gene names,
the sequences corresponding to the CDR1-IMGT, FR1-IMGT, CDR2-IMGT, FR2-IMGT,
CDR3-IMGT, FR3-IMGT and V-RS (recombination signal including V-HEPTAMER, V-SPACER
and V-NONAMER in 3' of V-REGION of a V-GENE) when available.
Simply select the labels to be shown using the popup menu at the bottom of the page.
- Note that the CDR-IMGT are omitted when the distance matrix is computed, they are still stored for later use. The same applies to the V-RS, which were
stored at selection time. For this reason, they can now be added to the tree representation.
- This kind of representation may yield interesting clues regarding the evolution of certain regions of the selected sequences.
For example, in the following screenshot, where the CDR1-IMGT are represented in addition to the tree computed from the mouse IGHV1 sequences,
the length of CDR1-IMGT nearly doubled (or was divided by two) in mouse sequences just after human-mouse speciation.
See also
References
-
[1] Elemento O and Lefranc M-P.
IMGT/PhyloGene, an online software package for phylogenetic analysis of immunoglobulin and T cell receptor genes.
Dev. Comp. Immunol., 27, 763-779 (2003),
PMID:12818634,
LIGM:273.
-
[2] Elemento O.
Phylogénie de familles multigéniques: Immunoglobulines et récepteurs T dans IMGT.
Thèse de doctorat, Université Montpellier II, 2002.
-
[3] Lefranc M-P.
IMGT, the international ImMunoGeneTics database®.
Nucleic Acids Res., 31, 307-310 (2003),
PMID:12520009,
LIGM:268.
-
[4] Kimura M.
A simple model for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences.
J. Mol. Evol., 16, 111-120 (1980),
PMID:7463489.
-
[5] Saitou N and Nei M.
The neighbor-joining method: a new method for reconstructing phylogenetic trees.
Mol. Biol. Evol., 4, 1073-1095 (1987),
PMID:3447015.
-
[6] Felsenstein J.
PHYLIP - PHYLogeny Inference Package.
Cladistics, 5, 164-166 (1989).
-
[7] Li W.
Molecular Evolution.
Sinauer Inc. Sunderland, Massachussets (1997).
-
[8] Swofford D, Olsen P, Waddell P and Hillis D.
Molecular Systematics. Chapter Phylogenetic Inference,
Sinauer Inc. Sunderland, Massachussets, 407-514 (1996).
IMGT/PhyloGene implementation and developments
The IMGT/PhyloGene tool was implemented by Olivier Elemento (LIGM, Montpellier,
France, Ph.D. thesis, Université Montpellier II 02/12/2002 [1,2])
using Perl and C programs.
Céline Protat added C-DOMAIN sequences into the IMGT/PhyloGene database and adapted the IMGT/PhyloGene
tool to deal with them in July 2003.
The IMGT/Phylogene tool has been improved by François Ehrenmann in October 2006.
- Created:
- 10/02/2002
- Last updated:
- 01/10/2006
- Authors:
- Olivier Elemento, François Ehrenmann, Céline Protat and Marie-Paule Lefranc
- Contact: Marie-Paule Lefranc
Marie-Paule.Lefranc@igh.cnrs.fr
- Editors:
- Quentin Kaas (2001-2007), François Ehrenmann (2006-2011),
Caroline Tournier (2011-2013), Typhaine Paysan-Lafosse (2013-2016)
and Patrice Duroux (LIGM, Montpellier, France)




{kind=link}