
| IMGT Web resources |
|
| Here you are: IMGT Web resources > IMGT Education > Tutorials > Cancer |
Les récepteurs à activité tyrosine kinase (RTKs) réarrangés dans les
hémopathies malignes
Géraldine GUASCH
U119 INSERM Laboratoire d'Oncologie Moléculaire
27 Bd Leï Roure 13009 Marseille
SOMMAIRE
1. Les récepteurs à activité tyrosine kinase (RTKs), hors FGFR
1-1. Structure et mécanisme d’activation des RTKs
1-2. Dérégulation des récepteurs à activité tyrosine kinase
1-2-1. Mutations ponctuelles de type gain-de-fonction, délétions partielles dans la séquence codante et mutations inhibitrices
1-2-2. Surexpression résultant d’une amplification génique
1-2-3. Réarrangements chromosomiques et formation d’une protéine de fusion
2. La famille des récepteurs des facteurs de croissance du fibroblaste: les FGFRs
2-1. Structure et mode d’activation des FGFRs
2-1-1. Les récepteurs à activité tyrosine kinase: FGFR1 à FGFR4
2-1-2. Le récepteur FGFRL1/FGFR5
2-2. Les isoformes des protéines FGFRs générées par épissage alternatif
2-3. Les voies de transduction associées à FGFR1: un réseau à multiples relais
2-3-1. Les protéines adaptatrices associées au récepteur FGFR1 activé
2-3-2. Les protéines à activité enzymatique activées par le récepteur FGFR1
2-4. Dérégulation des FGFRs
2-4-1. Mutations ponctuelles dans les FGFRs
2-4-2. Amplification du gène
2-4-3. Réarrangements chromosomiques
3. Références bibliographiques
Les récepteurs des facteurs de croissance à la surface des cellules constituent un groupe de molécules capables de recevoir des signaux de l'extérieur via leur partie extracellulaire pour les transmettre à l'intérieur de la cellule et induire de nombreux messages cellulaires. Ces récepteurs ont généralement une activité tyrosine kinase dans leur domaine intracellulaire. La transmission des messages à la cellule passe alors par l'autophosphorylation du récepteur sur résidus tyrosine. Les récepteurs à activité tyrosine kinase sont fréquemment dérégulés, soient par des mutations ponctuelles activatrices, soient par des translocations chromosomiques. Dans la troisième partie je détaillerai les récepteurs à activité tyrosine kinase fréquemment réarrangés dans les hémopathies malignes. La famille des récepteurs au facteur de croissance du fibroblaste, les FGFRs , sera plus particulièrement détaillée, puisque mon travail de thèse a porté sur l'étude de protéines FGFRs dérégulées suite à des translocations chromosomiques.
1. Les récepteurs à activité tyrosine kinase (RTKs), hors FGFR
A l'heure actuelle, il existe environ 58 récepteurs à activité tyrosine kinase. Selon leur organisation structurale, ces récepteurs se regroupent en plusieurs familles: 20 familles de RTKs ont été décrites (Figure 13) (Blume-Jensen et al., 2001 pour revue). Ces récepteurs ont une structure et des mécanismes d'activation assez similaires.
1-1. Structure et mécanismes d'activation des RTKs
Schématiquement, les RTKs se caractérisent par une région extracellulaire comportant le site de fixation du ligand (facteur de croissance pour la cellule) reliée par une unique région transmembranaire (TM) à une partie intracytoplasmique douée d'une activité enzymatique de phosphorylation sur des résidus tyrosine.
La partie extracellulaire comprend des domaines permettant au récepteur de dimériser (domaines de type immunoglobuline, fibronectine, domaines riches en cystéine, en leucine).
Le domaine transmembranaire est caractérisé par une séquence hydrophobe dont la fonction est l'ancrage du récepteur à la membrane. L'échange entre différents récepteurs de ces séquences d'acides aminés, a montré que l'origine du domaine transmembranaire est sans influence sur les capacités de signalisation des RTKs (Ullrich et al., 1990).
Le domaine intracytoplasmique tyrosine kinase est la partie la plus conservée parmi les RTKs. Il est formé de deux parties: une partie N-terminale capable de fixer l'ATP, complexé aux ions Mn2+ ou Mg2+, et une partie C-terminale dotée d'une activité phosphotransférase. L'activité kinase, résultant de l'autophosphorylation sur les résidus tyrosine dans le domaine catalytique, n'a aucun effet sur l'expression et la localisation des récepteurs à la surface des cellules. Cependant elle est cruciale pour l'activation des voies de transduction et pour l'induction des réponses cellulaires, comme la survie, la prolifération et la différenciation (Hubbard et al., 2000).
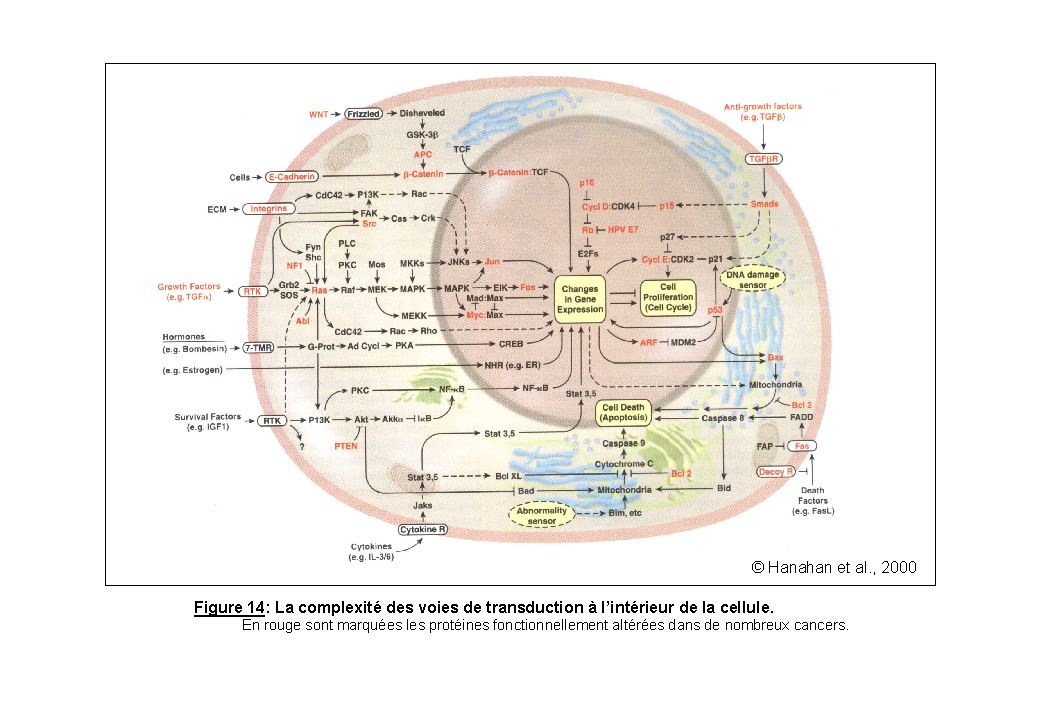
L'activation des RTKs est régulée suivant un processus commun. Les kinases possèdent une séquence appelée "boucle d'activation" dont le positionnement par rapport au site de fixation de l'ATP, conditionne l'activité du récepteur. La boucle d'activation doit être phosphorylée pour que la kinase soit active. Dans la conformation dite "ouverte", il y a fixation de l'ATP et transphosphorylation. Dans la conformation dite "fermée", la boucle masque le site de liaison de l'ATP, réduisant ainsi l'activité catalytique du récepteur. Ce mécanisme d'auto-inhibition, commun à de nombreuses kinases (Hubbard et al., 1998) est fréquemment perturbé dans les cancers. La répression normale des domaines catalytiques est alors inhibée formant des protéines constitutivement actives. Considérant la complexité des cascades de transduction à l'intérieur d'une cellule avec les nombreuses connexions entre les différentes voies (Figure 14), il est clair que la dérégulation de l'activation d'un RTK peut avoir de graves conséquences sur les réponses biologiques finales.
1-2. Dérégulation des récepteurs à activité tyrosine kinase
La transformation oncogénique induite par un RTK peut être déclenchée par plusieurs mécanismes: des mutations ponctuelles, ou des délétions dans le récepteur, une amplification génique ou des réarrangements chromosomiques. Ces altérations dans le récepteur peuvent être activatrices et aboutir soit à une activité kinase augmentée soit à une activité kinase constitutive (pour revue Blume-Jensen et al., 2001).
Les mutations peuvent toucher les différents parties du récepteur. Dans le cas de ERBB2/neu, récepteur du facteur de croissance épidermique (EGF), la mutation d'une valine en acide glutamique dans le domaine transmembranaire engendre l'activation du récepteur et l'acquisition d'un pouvoir oncogénique en favorisant probablement son oligomérisation (Weiner et al., 1989). Un autre exemple est retrouvé dans un cancer papillaire rénal où des mutations situées dans le domaine kinase du récepteur MET, récepteur du facteur de croissance de l'hépatocyte (HGF), engendrent une activité kinase constitutive (Schmidt et al., 1997; Jeffers et al., 1997). Ces mutations sont adjacentes à un domaine riche en glycine responsable de la fixation de l'ATP. Des mutations dans le domaine kinase de KIT, un membre de la famille des PDGFRs ont été identifiées chez des patients atteints de mastocytose, un syndrome caractérisé par une prolifération anormale des mastocytes (Longley et al., 1999). Il en résulte une activation constitutive du récepteur. Diverses mutations gain-de-fonction sont retrouvées dans le récepteur RET/GDNFR (Glial-Derived Neurotrophic Factor receptor): des mutations somatiques dans des tumeurs sporadiques et des mutations germinales trouvées dans trois syndromes familiaux qui sont des carcinomes médullaires de la thyroïde (MEN2A, MEN2B: Multiple endocrine neoplasia et FTMC: Familial medullary thyroid carcinoma) (Jhiang 2000). Les patients, présentant les syndromes MEN2A et FTMC, ont des mutations dans l'un des résidus cystéine présents dans le domaine extracellulaire de RET (Mulligan et al., 1993; Donis-Keller et al., 1993; Mulligan et al., 1994). La conséquence est la formation de ponts disulfure intramoléculaires entre des molécules RET, la dimérisation et l'activation constitutive du récepteur (Santoro et al., 1995). Dans le cas du syndrome MEN2B, la mutation se situe dans le domaine tyrosine kinase de RET, augmentant ainsi l'activité du récepteur sans induire la formation de dimères (Carlson et al., 1994; Hofstra et al., 1994). La mutation dans le domaine kinase altère également la spécificité des substrats de RET. Une augmentation de l'activité PI3Kinase, par exemple, est associée au syndrome MEN2B et doit participer au mécanisme de transformation maligne (Murakami et al., 1999).
Des délétions peuvent également être activatrices pour le récepteur. Dans le cas de ERBB1, une délétion dans la partie extracellulaire du récepteur est retrouvée dans de nombreux cancers (cancer du poumon, du sein, de l'ovaire, des gliomes). Cette délétion maintient le récepteur en permanence dans une conformation active (Moscatello et al., 1995).
Il existe également des mutations qui inhibent l'activité kinase du récepteur. C'est le cas par exemple des mutations A621-T, H650-P et F584-L qui touchent des résidus dans le domaine kinase de KIT très conservés parmi tous les RTKs (Spritz et al., 1992; Ezoe et al., 1995). Ces mutations dans KIT sont responsables du piebaldisme, une maladie génétique autosomique dominante qui se caractérise par un défaut de pigmentation de la peau.
1-2-2. Surexpression résultant d'une amplification génique
Beaucoup d'exemples de RTKs surexprimés dans les cancers existent et ont été répertoriés par Blume-Jensen et al., (2001). La surexpression des RTKs aboutit à l'activation constitutive de la kinase en augmentant la concentration des dimères. Un exemple très décrit dans la littérature est celui de ERBB2/neu qui est fréquemment surexprimé dans des cancers du sein, de l'estomac et du poumon. L'amplification de ERBB2 dans les cancers du sein a une valeur de pronostic (Slamon et al., 1987; Yarden et al., 2001 pour revue). ERBB1 et PDGFRb sont surexprimés dans des tumeurs humaines gliales (Fleming et al., 1992). Un autre exemple est celui du récepteur MET surexprimé dans de nombreux types de carcinomes humains (Beviglia et al., 1997). Cette surexpression suggère que le récepteur MET est impliqué dans le développement et la progression des tumeurs épithéliales. De nombreux sarcomes surexpriment également MET (Wallenius et al., 2000), comme des sarcomes de rat (Helou et al., 1997) et des ostéosarcomes humains (Ferracini et al., 1995). Il semble que la surexpression de MET et/ou de son ligand aboutisse à une boucle d'activation autocrine, à l'origine de l'activation de MET (Ferracini et al., 1995). Un autre récepteur KIT est surexprimé dans des carcinomes de glandes salivaires (Jeng et al., 2000). Chez des patients atteints de mastocytose, l'expression de l'ARNm de KIT est élevée (Nagata et al., 1998).
1-2-3. Réarrangements chromosomiques et formation d'une protéine de fusion
Les protéines de fusion impliquant les protéines à activité tyrosine kinase, sont généralement composées d'un domaine d'oligomérisation en N-terminal fusionné à un domaine catalytique. Un mécanisme commun d'activation de ces protéines de fusion peut être décrit dans lequel le domaine d'oligomérisation est responsable de la dimérisation et par conséquence de l'activation constitutive de la kinase. De nombreux exemples de RTKs dérégulés par des aberrations chromosomiques ont été décrits dans les cancers solides et les hémopathies malignes (Blume-Jensen et al., 2001).
Le récepteur RET est impliqué dans au moins huit réarrangements chromosomiques (inversions et translocations) associés à des carcinomes papillaires thyroïdiens. Les aberrations chromosomiques fusionnent la partie N-terminale d'une protéine avec le domaine tyrosine kinase de RET (Jhiang 2000). Le récepteur TRKA, dans des carcinomes papillaires thyroïdiens et des neuroblastomes, est également réarrangé dans au moins trois translocations chromosomiques qui créent des protéines de fusion ayant le domaine tyrosine kinase de TRKA en C-terminal (Greco et al., 1992; Greco et al., 1995; Butti et al., 1995). Enfin les récepteurs TRKC, PDGFRb, sont dérégulés dans les translocations t(12;15) et t(5;12). Ces translocations fusionnent le domaine d'oligomérisation de TEL avec le domaine tyrosine kinase des RTKs. La protéine de fusion TEL-TRKC est associée au fibrosarcome congénital et à des leucémies aiguës myéloïdes (Knezevich et al., 1998) et la protéine TEL-PDGFRb est retrouvée dans des leucémies chroniques myélomonocytaires (Golub et al., 1994).
2. La famille des récepteurs des facteurs de croissance du fibroblaste: les FGFRs
2-1. Structure et mode d'activation des FGFRs
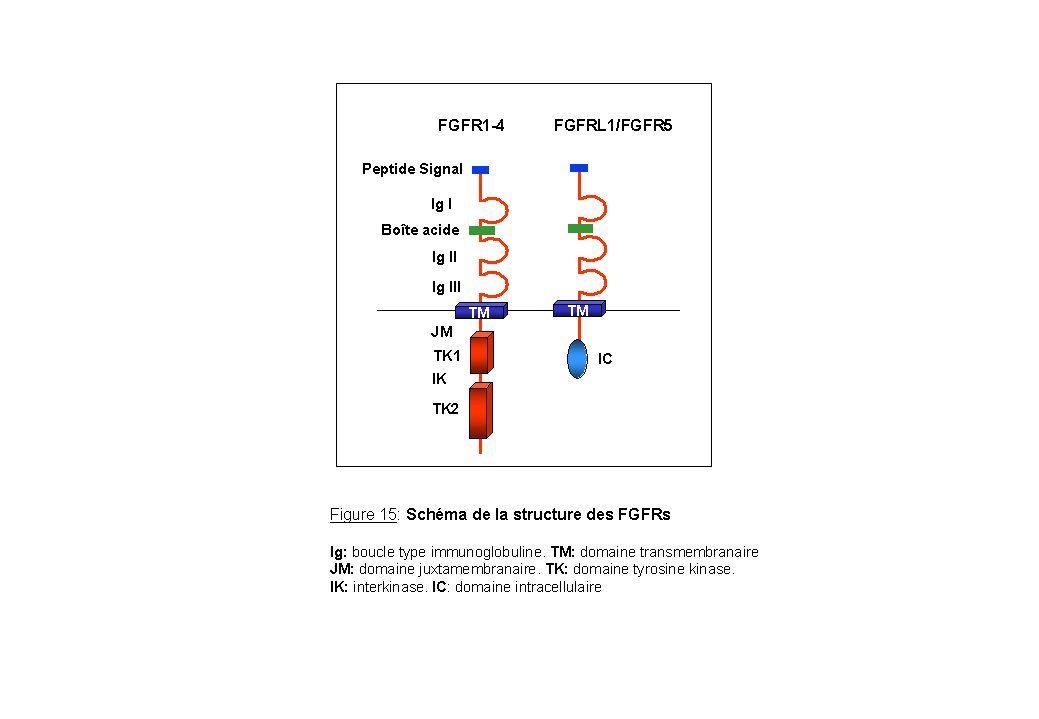
La famille des FGFRs, appartient à la classe IV des RTKs (Figure 13) (Ullrich et al., 1990; Johnson et al., 1991; Wilks 1993; Heldin 1996). Elle comprend chez les mammifères quatre membres (FGFR1-4) (Dionne et al., 1990) présentant de fortes identités de séquences, allant de 55 à 70% (Johnson et al., 1993). Des gènes orthologues aux FGFRs ont été caractérisés chez les non vertébrés : un chez C.elegans nommé egl-15 et deux chez la drosophile D. Melanogaster nommés Heartless et Breathless (DeVore et al., 1995; Beiman et al., 1996; Coulier et al., 1997). Récemment il a été découvert un nouveau membre de la famille des FGFRs, pouvant être considéré comme une sous-classe de FGFR. Deux nomenclatures ont été données pour ce même récepteur: FGFRL1 (Wiedemann et al., 2000) et FGFR5 (Kim et al., 2001; Sleeman et al., 2001) (Figure 15).
Les FGFRs sont les récepteurs des facteurs de croissance des fibroblastes (FGF).
Les FGF sont des protéines monomériques de 17 à 34 kDa regroupées au sein d'une famille multigénique formée à ce jour de 22 facteurs (Ornitz et al., 2001). Les FGF sont composés d'une région centrale dite "core", fortement conservée entre les différents FGF, qui permet la liaison aux récepteurs de haute affinité (les FGFRs) et aux récepteurs de basse affinité (les protéoglycanes) (Goldfarb 1990). La majorité des FGF possèdent un peptide signal en N-terminal facilitant leur sécrétion vers le milieu extracellulaire. Ils sont parfois internalisés avec une localisation nucléaire ou nucléolaire (Arese et al., 1999). Chaque FGF peut se lier à plusieurs FGFRs et réciproquement, chaque FGFR peut lier plusieurs FGF.
2-1-1. Les récepteurs à activité tyrosine kinase: FGFR1 à FGFR4
Ces protéines dont la taille varie entre 110 et 150 kDa, sont constituées de la façon suivante: (Figure 15).
Ø une région N-terminale extracellulaire. Elle comporte un peptide signal nécessaire pour le transport de la protéine à la membrane, deux ou trois boucles de type immunoglobuline (Ig) et une région acide, constituée de 4 à 8 acides aspartiques ou glutamiques, située entre la première et la deuxième boucle Ig. Cette boîte acide semble cruciale pour la fonction du récepteur : en effet, sa délétion engendre une complète inhibition de la fixation du ligand sur son récepteur (Chaudhuri et al., 1993).
La structure cristallographique de la région extracellulaire de FGFR1 complexée avec son ligand a montré que FGF2 interagit avec des dimères de récepteurs et qu'il n'y a pas d'interaction directe entre deux molécules de FGF (Plotnikov et al., 1999). Ceci a été confirmé sur les complexes FGF1-FGFR2 (Stauber et al., 2000), FGF1-FGFR1 et FGF2-FGFR2 (Plotnikov et al., 2000) montrant qu'il existe un mécanisme commun dans l'activation des FGFRs.
La fixation du ligand FGF sur son récepteur induit le changement conformationnel par dimérisation et par la suite l'activation du récepteur (Schlessinger et al., 1992). Les FGF sont des ligands monomériques qui doivent être préalablement dimérisés pour se lier aux récepteurs. La dimérisation des FGF est facilitée par l'interaction avec des glycoprotéines spécifiques: l'héparine et les sulfates d'héparane (Yayon et al., 1991; Spivak-Kroizman et al., 1994). Les sulfates d'héparane sont un des constituants de la matrice extracellulaire, sous la forme de protéoglycanes riches en sulfate d'héparane (HSPG), et jouent un rôle majeur dans la localisation des FGF au niveau de la matrice extracellulaire (pour revue Klagsbrun et al., 1991). La liaison spécifique du ligand se fait au niveau de la seconde et de la troisième boucle Ig ainsi qu'au niveau de la région comprise entre ces deux boucles (Jaye et al., 1992; Johnson and Williams 1993). La structure cristallographique du complexe ternaire FGF2-FGFR1-Héparine a mis en évidence un double rôle pour l'héparine: elle augmente l'affinité du FGF envers le FGFR et stabilise le dimère (Schlessinger et al., 2000c).
Ø une région C-terminale intracellulaire séparée de la partie extracellulaire par un domaine transmembranaire. Elle comprend un domaine catalytique composé de deux sous-domaines tyrosine kinase, TK1 et TK2, interrompu par une courte séquence interkinase de 14 acides aminés. Cette région cytoplasmique comprend au moins 7 tyrosines qui sont des sites d'autophosphorylation (Mohammadi et al., 1996a). Dans la conformation dite "fermée", la boucle d'activation est située dans le site catalytique et inhibe la fixation des substrats de la protéine (Mohammadi et al., 1996b). L'activité kinase des FGFRs est cruciale pour l'activation des voies de transduction et pour l'induction des réponses cellulaires comme la mitogenèse ou la différenciation (Mohammadi et al., 1996a). Cependant, les FGFRs ont des potentiels mitogéniques différents et le niveau d'activation des voies de transduction varie donc d'un FGFR à l'autre (Wang et al., 1994). FGFR4 par exemple, induit peu de réponses mitogéniques contrairement à FGFR1 et FGFR3. En effet, des cellules hématopoïétiques stimulées par le FGF, surexprimant les récepteurs FGFR1, FGFR2 ou FGFR3 sont capables de proliférer en absence de cytokine à l'inverse de FGFR4 qui est incapable de promouvoir la prolifération dans ces cellules (Ornitz et al., 1992; Wang et al., 1994). La différence dans le potentiel mitogénique des FGFRs vient du domaine kinase. En effet, une construction chimère formée du domaine extracellulaire de FGFR4 fusionné au domaine kinase de FGFR1 est capable, comme FGFR1, d'induire la prolifération des cellules hématopoïétiques en absence de cytokine (Wang et al., 1994). Les mêmes résultats ont été obtenus dans des myoblastes de rat où seuls FGFR1 et FGFR2 sont capables de former des colonies en agar (Shaoul et al., 1995). Le récepteur FGFR1 a été le plus étudié tant au niveau cristallographie qu'au niveau des voies de transduction activées par la fixation des FGF.
La structure cristallographique du domaine kinase de FGFR1 a permis de comprendre les mécanismes d'action des RTKs (Mohammadi et al., 1996b). Elle a permis de visualiser les sites d'autophosphorylation et de comprendre comment diverses mutations dans la partie kinase aboutissent à la formation de désordres du développement (pour revue Roberston et al., 2000).
2-1-2. Le récepteur FGFRL1/FGFR5
FGFRL1/FGFR5 est un récepteur transmembranaire qui comporte une région extracellulaire similaire aux protéines FGFR3 et FGFR4 composée de 3 boucles de type immunoglobuline et d'une boîte acide (Wiedemann et al., 2000). Le domaine transmembranaire de FGFRL1/FGFR5 est également conservé avec notamment le résidu glycine G380 crucial dans FGFR3 à la même position: la mutation de ce résidu glycine en arginine dans FGFR3 est responsable de l'achondroplasie (voir tableau 2). Le domaine intracellulaire par contre est dépourvu de domaine tyrosine kinase et ne ressemble à aucune autre séquence protéique retrouvée dans les banques de données. FGFRL1/FGFR5 ne peut donc pas être répertorié dans la classe IV des RTKs. La fixation du ligand sur ce récepteur reste encore à être démontré. Cependant, la partie cytoplasmique comporte deux résidus tyrosine intégrés au niveau de motifs consensus de liaison aux protéines tyrosine phosphatase SHP. La présence d'un tel motif suggère que FGFRL1/FGFR5 est capable de transmettre des signaux moléculaires suite à la fixation du ligand FGF.
Cette nouvelle protéine, apparentée aux FGFRs, est exprimée dans le pancréas, le foie, le rein, le coeur, le muscle squelettique et le cartilage. Dans ce dernier, elle pourrait moduler l'activité de FGFR3 également exprimé dans ce tissu et jouer un rôle dans la différenciation des chondrocytes et dans l'ossification endochondrale. Un rôle possible pour FGFRL1/FGFR5 pourrait être d'interagir avec d'autres FGFRs et d'inhiber la signalisation en bloquant la transphosphorylation du récepteur. Une protéine agissant ainsi a été décrite dans la signalisation associée au TGFb: BAMBI (BMP and activin membrane- bound inhibitor), une protéine transmembranaire, apparentée au TGFbRI dépourvue de domaine intracellulaire serine/thréonine kinase. Ce pseudo-récepteur s'associe au TGFbR et inhibe la voie de signalisation associée (Onichtchouk et al., 1999)
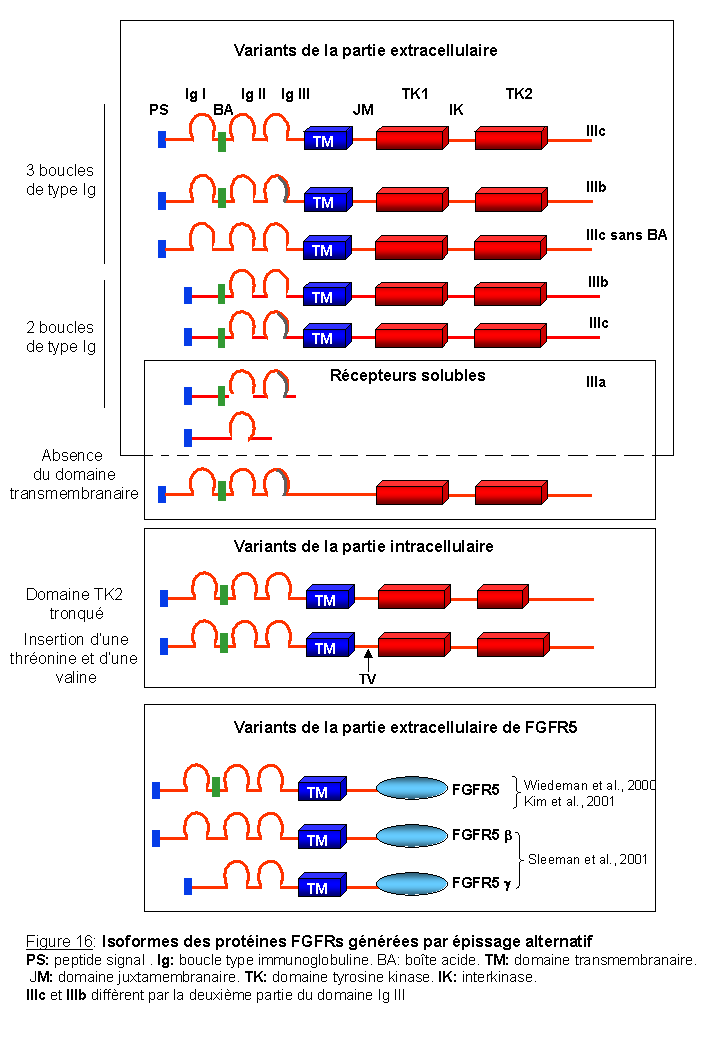
2-2. Les isoformes des protéines FGFRs générées par épissage alternatif
Les FGFRs existent sous de nombreuses isoformes (Figure 16) qui résultent d'un épissage alternatif. Les sites d'épissage alternatif sont essentiellement localisés dans la partie extracellulaire du récepteur, au niveau des domaines de type immunoglobuline: isoformes avec deux ou trois boucles de type immunoglobuline, isoformes différentes par la deuxième partie du dernier domaine immunoglobuline (trois exons différents: formes appelées IIIa, IIIb, et IIIc), ou isoformes constituées exclusivement de la partie extracellulaire (formes sécrétées). Il existe également des isoformes différentes par la partie intracellulaire du récepteur. L'épissage alternatif se situe au niveau du domaine juxtamembranaire et résulte en l'insertion de deux acides aminés, valine et thréonine (Twigg et al., 1998). Ces acides aminés dans FGFR1 sont cruciaux pour l'activation des voies de signalisation MAPK (Burgar et al., 2001). Les isoformes produites permettent ainsi l'obtention d'un très grand nombre de combinaisons qui confèrent une spécificité de liaison pour les FGF. L'isoforme FGFR2 IIIb, qui comprend deux domaines de type immunoglobuline dans sa partie extracellulaire en l'absence de la boîte acide, code pour le récepteur KGFR, récepteur du facteur de croissance du kératinocyte (Battaro et al., 1990; Miki et al., 1991). Le KGFR lie avec haute affinité FGF1, FGF7/KGF et FGF10.
Dans FGFR5, c'est la partie extracellulaire de ce récepteur qui est la plus conservée par rapport aux autres FGFRs. Comme pour les autres récepteurs, des sites d'épissage alternatifs, situés au niveau des séquences codantes de la première boucle immunoglobuline, permettent de créer deux variants: FGFR5b et FGFR5g (Sleeman et al., 2001).
2-3. Les voies de transduction associées à FGFR1: un réseau à multiples relais
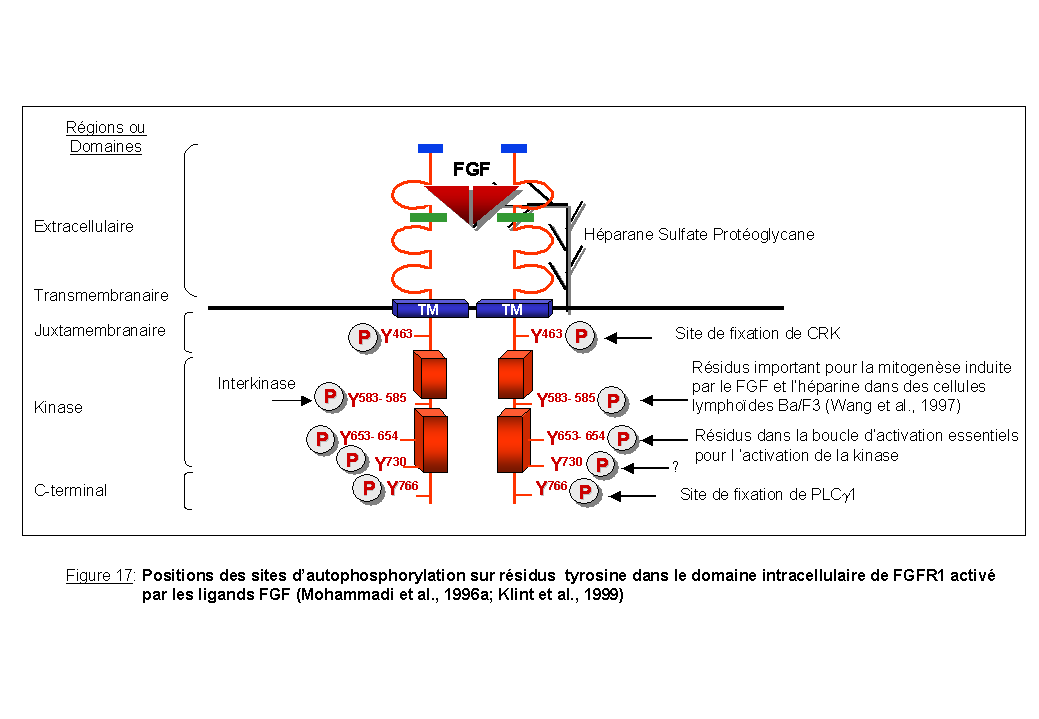
Les RTKs transmettent des messages à la cellule suite à leur autophosphorylation sur résidus tyrosine. Les sites d'autophosphorylation de la protéine FGFR1 activée suite à la fixation du ligand FGF complexé à des héparanes sulfate protéoglycanes ont été mis en évidence par Mohammadi et al., (1996a) (Figure 17). Les informations concernant les voies activées par les récepteurs FGFRs et leurs gènes cibles dans les cellules du système hématopoïétique sont peu nombreuses et incomplètes (Wang et al., 1994; Klint et al., 1999). Il a été montré que FGFR1 stimule la prolifération cellulaire et est impliqué dans des mécanismes de transformation cellulaire. En effet, des cellules lymphoïdes Ba/F3 (dépendantes de l'IL-3 pour leur survie et leur prolifération) surexprimant à leur surface le récepteur FGFR1, sont capables de proliférer en absence d'IL-3 en réponse au FGF (Wang et al., 1994). De plus, Wang and Goldfarb (1997) ont montré que les résidus tyrosine Y583 et Y585 présents dans le domaine interkinase de FGFR1 permettent la transmission d'un fort signal mitogénique. Certains sites comme les tyrosines Y653 et Y654 présents dans la boucle d'activation sont cruciaux pour l'activité kinase du récepteur.
L'autophosphorylation du récepteur a deux conséquences:
1. elle stimule l'activité catalytique intrinsèque du récepteur
2. elle génère des tyrosines phosphorylées qui représentent alors des sites pour la reconnaissance et le recrutement de protéines nécessaires à la transduction du signal sous les RTKs. Ces protéines en aval du récepteur contiennent des domaines de reconnaissance des tyrosines phosphorylées tels que le domaine SH2 (Src Homology 2) ou le domaine PTB (PhosphoTyrosine Binding) (Pawson et al., 1997; Broutin et al., 2000; Pawson et al., 2000; Schlessinger 2000a) (http://smart.embl-heidel-berg.de/ pour les domaines structuraux retrouvés dans les protéines de la transduction du signal).
Les différents substrats de FGFR1 peuvent être classés en deux catégories: les protéines dites "adaptatrices" et les protéines ayant une activité enzymatique (phospholipase pour la PLCg1, GTPase pour RAS, tyrosine kinase pour SRC, protéine kinase pour AKT/PKB, phosphatase pour SHP2)
2-3-1. Les protéines adaptatrices associées au récepteur FGFR1 activé (Tableau 1)
Les protéines adaptatrices, associées au récepteur FGFR1, présentent des domaines SH2 et SH3 et sont dépourvues de toute activité catalytique. Quatre ont été identifiées: CRK, GRB2, FRS2/SNT et SHC.
CRK (CT10 regulator of kinase: oncogène du virus CT10) est une protéine adaptatrice qui contient un domaine SH2 et deux domaines SH3 (Mayer et al., 1988; Feller 2001 pour revue). Les domaines SH3 lient des motifs PXXP riches en proline et sont impliqués dans les interactions protéine- protéine. CRK s'associe via son domaine SH2 à la tyrosine 463 phosphorylée dans le domaine juxtamembranaire de FGFR1 (Figure 17 et Figure 18). Des cellules surexprimant un récepteur FGFR1 muté au niveau de cette tyrosine (Y463/F), sont incapables de proliférer en présence du ligand FGF. Cette inhibition de la prolifération est corrélée avec une réduction de l'activation de la voie MAPK et une absence de phosphorylation d'une protéine FRS2/SNT, décrite ci-dessous, impliquée dans l'activation de la voie RAS/MAPK. La surexpression d'une forme mutée de CRK dans le domaine SH2 a les même effets sur la prolifération des cellules que le mutant FGFR1 Y463/F.
Ces expériences montrent que la fixation et la phosphorylation de CRK sont des événements importants pour la prolifération induite par le couple FGF-FGFR1 (Larsson et al., 1999). Récemment, des études ont mis en évidence un rôle de CRK dans la survie cellulaire. En effet, CRK induit la transformation de fibroblastes embryonnaire de poulet et de fibroblastes NIH3T3 en activant de façon constitutive la voie PI3Kinase/AKT (décrite ci-dessous) (Akagi et al., 2000; Stam et al., 2001).
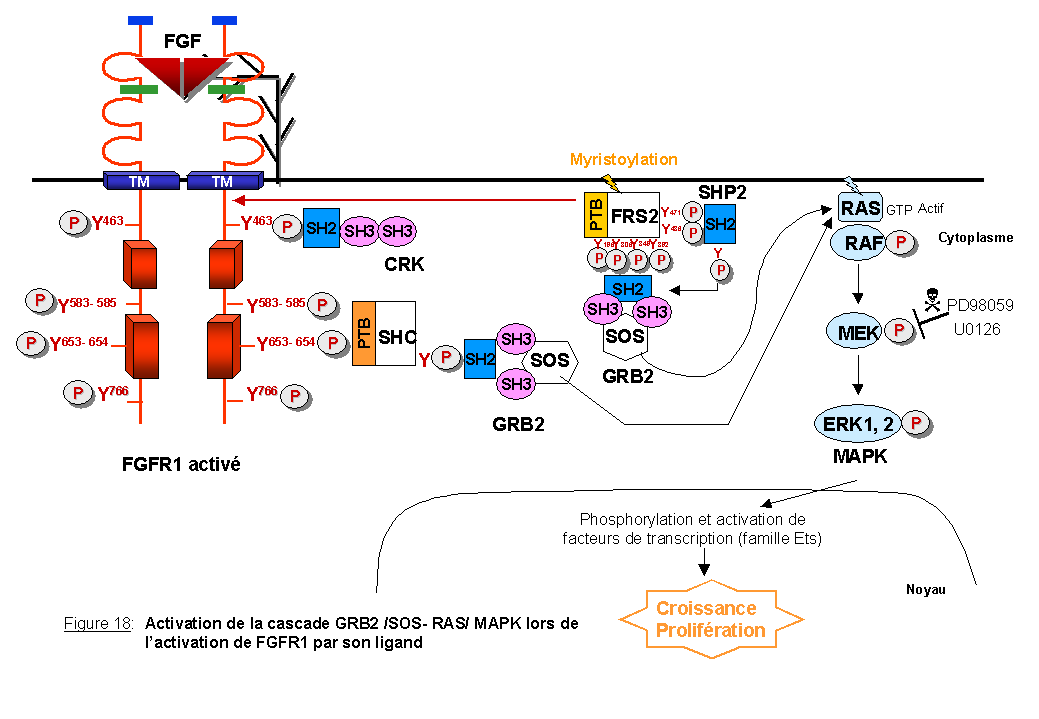
FGFR1 est capable de recruter de façon indirecte GRB2 (Growth factor receptor-bound protein 2), une protéine adaptatrice composée de deux domaines SH3 encadrant un domaine SH2. Elle permet aux récepteurs d'activer la voie RAS/MAPK (Lowenstein et al., 1992) (Figure 18). La protéine GRB2 forme un complexe avec SOS (Son on Sevenless), le facteur d'échange de la guanine, via son domaine SH3. SOS catalyse l'échange de GDP en GTP sur une petite protéine G RAS, localisée à la membrane plasmique, permettant ainsi son activation sous forme RAS-GTP (Shannon 1995). RAS sous forme activée se fixe sur la protéine RAF, une MAP Kinase- kinase- kinase. L'interaction RAS-GTP- RAF stimule une série de kinases qui induisent un signal de croissance et de prolifération dans le noyau (Chang et al., 2001 pour revue sur les cascades MAP kinase) (Figure 18). Les récepteurs des FGF sont dépourvus du site, Y-X-N, de fixation directe à GRB2 (Songyang et al., 1993). Il existe donc des protéines intermédiaires entre le récepteur FGFR1 et le complexe GRB2-SOS. FRS2/SNT et SHC sont deux protéines intermédiaires phosphorylées sur tyrosine par FGFR1. Les tyrosines phosphorylées de ces deux protéines représentent des sites de fixation pour le domaine SH2 de GRB2 (Klint et al., 1995).
FRS2/SNT: FRS2 murin (pour FGF receptor substrate 2) appartient à une famille de protéines aussi nommée SNT (Suc-1-associated Neurotrophic factor Target) (Wang et al., 1996). SNT1 est l'homologue humain de FRS2. FRS2 est phosphorylée sur tyrosine par des récepteurs à activité tyrosine kinase stimulés par des facteurs, comme le FGF ou le NGF, capables d'engendrer la différenciation de cellules neuronales PC12 (Rabin et al., 1993; Wang et al., 1996). FRS2 est une molécule de 90 kDa myristoylée qui contient un domaine PTB dans sa partie NH2-terminale. L'interaction entre le domaine PTB de FRS2 et FGFR1 se fait au niveau des résidus 419-430 dans le domaine juxtamembranaire de FGFR1 où est retrouvée une séquence non phosphorylée, KSIPLRRQVTVS, fortement conservée parmi la famille des FGFRs de mammifères (Xu et al., 1998). Burgar et al., (2001) ont montré que les résidus valine et thréonine (VT) présents dans la partie juxtamembranaire de FGFR1, sont cruciaux pour l'interaction avec FRS2. Bien que cette séquence ne contienne pas les résidus asparagine et tyrosine retrouvés dans tous les motifs de reconnaissance des domaines PTB, elle permet à FRS2 de se fixer à FGFR1. Des variants dans les motifs de reconnaissance des domaines PTB ont déjà été décrits (Borg et al., 1996). En ce qui concerne le récepteur TRKA à la neurotrophine, le PTB de FRS2 se fixe au niveau de la séquence consensus phosphorylée NPXY. Donc en fonction du type de récepteur, le PTB de FRS2 peut fixer des séquences phosphorylées ou non phosphorylées.
La structure de FRS2 est unique mais ses fonctions miment celles d'une autre protéine IRS1 (Insulin Receptor Substrate 1). IRS1 contient un domaine PH "Pleckstrin Homology" (pouvant avoir les mêmes fonctions que la séquence de myristoylation de FRS2), et un domaine PTB (29% d'identité avec le PTB de FRS2 et 50% de similitude). Ce domaine fixe une tyrosine phosphorylée dans la séquence consensus NPXY-p de reconnaissance des domaines PTB, au niveau du domaine juxtamembranaire du récepteur à l'insuline (O'Neill et al., 1994; Yenush et al., 1997 pour revue).
Il a récemment été montré que l'interaction entre le domaine PTB de FRS2 et le domaine juxtamembranaire de FGFR1 est indépendante de l'activation du récepteur: en effet, un récepteur FGFR1 n'ayant plus d'activité kinase coimmunoprécipite FRS2 de la même façon qu'un récepteur FGFR1 sauvage (Ong et al., 2000). Donc FRS2 peut interagir directement avec FGFR1 en absence de toute stimulation du récepteur.
FRS2 est phosphorylée sur 4 tyrosines (Y196-306-349-392) en réponse au FGF ou au NGF et ces tyrosines phosphorylées sont des sites de fixation pour le domaine SH2 de GRB2 (Kouhara et al., 1997). Ceci aboutit à l'activation des voies RAS/MAPK engendrant la prolifération (Szeberenyi et al., 1990). La myristoylation de FRS2 est essentielle pour sa localisation membranaire, sa phosphorylation sur tyrosine, le recrutement de GRB2/SOS et l'activation de la voie MAPK.
FRS2 est également phosphorylée sur deux autres tyrosines (Y436 et 471) ce qui entraîne la fixation d'une phosphatase SHP2. SHP2 est à son tour phosphorylée, fixe le complexe GRB2/SOS et active la voie RAS/MAPK (Hadari et al., 1998). L'activité catalytique de SHP2 est essentielle pour produire un signal de prolifération et de différenciation. L'invalidation génique de FRS2, léthale au niveau embryonnaire, a récemment montré que cette protéine n'est pas nécessaire pour l'activation de FGFR1 mais est requise pour la phosphorylation de SHP2 et par conséquent importante pour la formation du complexe SHP2-GRB2 (Hadari et al., 2001).
SHC (SH2-containing sequence ou Src homologous and collagen) est une protéine qui existe sous trois isoformes. Deux d'entre-elles (52 kDa et 46 kDa) sont composées de trois domaines: un domaine SH2 situé en C-terminal, un domaine central CH1 (collagen homology 1) riche en résidus proline et glycine et un domaine PTB en N-terminal (Pelicci et al., 1992; Ravichandran 2001 pour revue). La troisième isoforme de 66 kDa, issue d'un épissage alternatif, possède en N-terminal un domaine supplémentaire CH2 (collagen homology 2) également riche en résidus proline et glycine (Migliaccio et al., 1997). Les trois isoformes de SHC sont capables, via leur domaine SH2 et/ou PTB, de se fixer aux résidus phosphorylés de nombreux récepteurs à activité tyrosine kinase tels que le récepteur à l'EGF, au NGF ou au PDGF pour être ensuite elles-mêmes phosphorylées (pour revue Bonfini et al., 1996).
L'activation du récepteur FGFR1 corrèle avec la phosphorylation de SHC sur résidu tyrosine. Le résidu tyrosine phosphorylé permet à son tour la liaison au domaine SH2 de GRB2 (Klint et al., 1995) (Figure 18). L'interaction directe entre FGFR1 et SHC n'a jamais été clairement démontrée in vivo, SHC ne possédant pas le site spécifique putatif de liaison pour le domaine SH2 (Klint et al., 1995). Il est possible que cette interaction soit trop "éphémère" pour être détectée ou qu'il existe une autre protéine adaptatrice entre FGFR1 et SHC. D'autres études ont montré l'absence d'association entre le complexe SHC-GRB2 et SOS, laissant suggérer que l'activation de SHC par FGFR1 ne doit pas activer directement la phosphorylation de ERK1 et ERK2 (Raffioni et al., 1999).
2-3-2. Les protéines à activité enzymatique activées par le récepteur FGFR1 (Tableau 1):
La transduction du signal intracellulaire à partir de FGFR1 activé est encore peu connue. L'activation de FGFR1, via le recrutement et la phosphorylation de divers substrats, activent des voies de transduction parallèles et induit de nombreux messages cellulaires comme la mitogenèse, la prolifération ou la survie, la différenciation, la migration et la mort cellulaire (Pour revue Klint et al., 1999). Il est connu que FGFR1 activé induit la survie cellulaire par la voie PI3Kinase, la mitogenèse et la prolifération par la voie PLCg1 qui active la voie RAS/MAPK.
FGFR1 et la voie PI3Kinase/AKT:
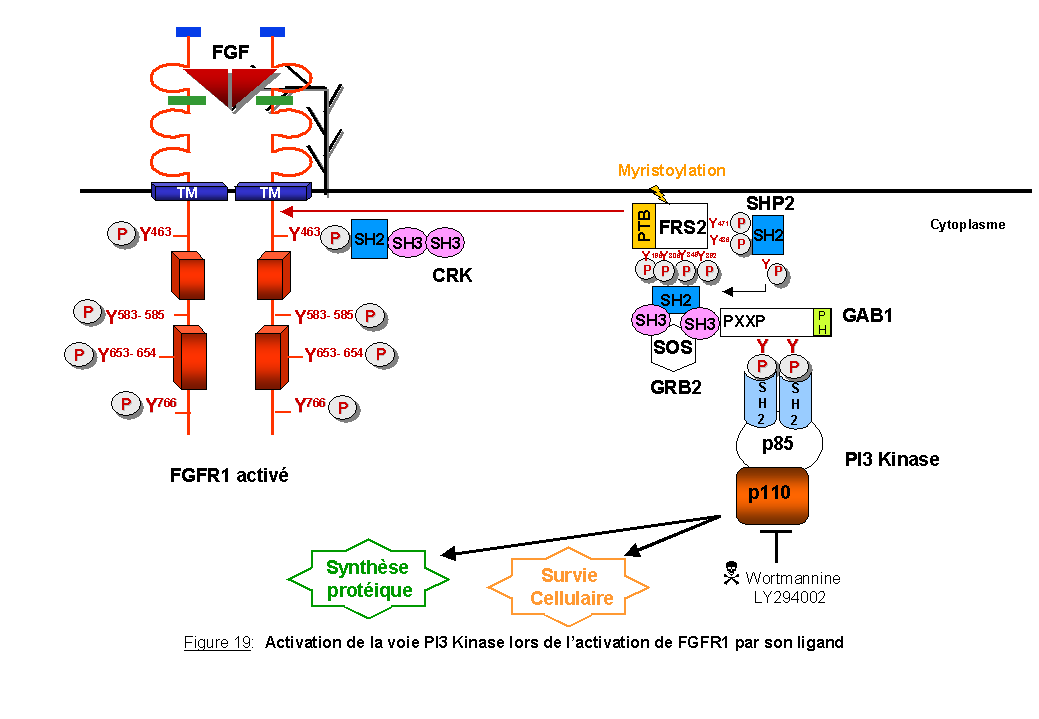
Nous venons de voir que FRS2/SNT et SHC, par des interactions avec diverses molécules, activent la voie RAS/MAPK. Des études récentes ont montré que FRS2 et SHC dans le système FGF-FGFR1 sont capables d'activer la voie phosphatidylinositol 3'-OH kinase (PI3Kinase) aboutissant à la survie cellulaire (Figure 19). Il a récemment été montré un rôle de SHC dans l'activation de la voie PI3K/AKT (Gu et al., 2000).
Certains récepteurs recrutent directement la PI3Kinase comme le PDGFR et ERBB3. D'autres utilisent des protéines intermédiaires comme IRS1 dans le cas du récepteur à l'insuline ou GAB1 dans le cas de MET (le récepteur du facteur de croissance de l'hépatocyte) (Fan et al., 2001) et de FGFR1 (Ong et al., 2001).
GAB1 (GRB2 Associated Binder) a été initialement cloné comme une protéine associée à GRB2 et phosphorylée sur tyrosine après stimulation des cellules par l'EGF ou l'insuline (Holgado- Madruga et al., 1996; Rodrigues et al., 2000). GAB1 partage de nombreuses similitudes en acides aminés avec IRS1 au niveau du domaine PH (Pleckstrin Homology) retrouvé dans les deux protéines (Yenush et al., 1997). Dans le système FGF-FGFR1, FRS2 forme un complexe avec GRB2 qui fixe, via ses domaines SH3, une région riche en proline de GAB1. GAB1 contient 3 résidus tyrosine dans des séquences consensus YXXM qui représentent des sites de fixation pour le domaine SH2 de la sous- unité p85 de la PI3Kinase (Ong et al., 2001) (Figure 19). Il en résulte une activation de la voie PI3Kinase.
Lors de l'induction du mésoderme chez l'embryon de xénope, il a été montré que la PI3Kinase et les ERK agissent en coopération dans la voie de signalisation induite par le couple FGF-FGFR (Carballada et al., 2001).
Des études portant sur la stimulation de la PI3Kinase par FGFR1 ont montré que la surexpression de FRS2 ou de GAB1 dans des fibroblastes NIH3T3 stimulés par FGF induit l'activation de AKT (Ong et al., 2001). En utilisant les oocytes de xénope exprimant de façon endogène FGFR1, Browaeys-Poly et al., (2000) ont également mis en évidence la phosphorylation de AKT après stimulation des oocytes par le FGF.
FGFR1 et la voie de la PLCg1:
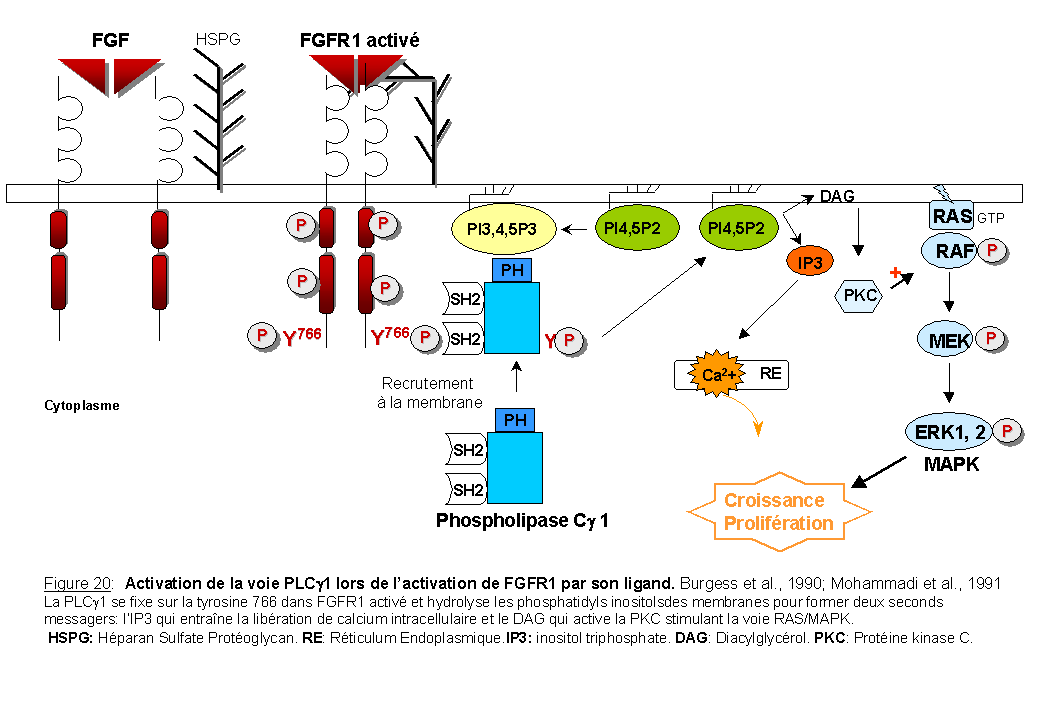
L'activation de la voie PI3Kinase est connectée à l'activation d'une autre enzyme la phospholipase C-g1( PLCg1) par l'intermédiaire des seconds messagers lipidiques (Falasca et al., 1998) et d'un site d'autophosphorylation, la tyrosine Y766 présente dans la région C-terminale de FGFR1 (Cross et al., 2000) (Figure 17). La PI3Kinase régule également la voie du calcium associée à la PLCg (Rameh et al., 1998; Barker et al., 1999).
La PLCg est une enzyme intracellulaire qui contient deux domaines SH2, un domaine SH3 et un domaine PH (pour revue Carpenter et al., 1999). La PLCg1 est phosphorylée suite à l'activation de FGFR1 (Burgess et al., 1990). FGFR1 présente une tyrosine (Y766) phosphorylée dans sa région C-terminale qui fixe le domaine SH2 de la PLCg1, et phosphoryle cet enzyme sur tyrosine: ceci aboutit à l'hydrolyse des phosphoinositides (PIP2) pour former deux seconds messagers: le diacylglycérol (DAG) et l'inositol triphosphate (IP3) (Mohammadi et al.,1991) (Figure 20). L'IP3 entraîne la libération du calcium intracellulaire et le DAG active la protéine kinase C qui phosphoryle RAF-1 et stimule la voie RAS/MAPK (Sozeri et al., 1992; Kolch et al., 1993).
Un récepteur FGFR1 muté sur cette tyrosine par substitution en un acide aminé phénylalanine Y766/F, après activation par le FGF, est incapable de fixer la PLCg, n'hydrolyse pas les phosphoinositides et ne libère pas le calcium intracellulaire. L'internalisation du récepteur semble également être perturbée (Sorokin et al., 1994). Cependant cette mutation n'affecte pas la mitogenèse des myoblastes L6 (synthèse d'ADN mesurée par incorporation de thymidine tritiée) (Mohammadi et al., 1992; Peters et al.,1992) ni la différenciation neuronale de cellules PC12 (Spivak-Kroizman et al., 1994b).
En accord avec ces résultats, Huang et al., (1995) ont montré que l'activation de RAF et de MAPK est diminuée dans des cellules Ba/F3 stimulées par FGF et surexprimant le récepteur FGFR1 Y766/F. La PLCg1 par l'intermédiaire de la PKC modifie donc l'état de phosphorylation de RAF et par conséquent son activité. La prolifération des cellules Ba/F3, exprimant le récepteur FGFR1 Y766/F, et cultivées en absence d'IL-3 mais en présence de FGF, est similaire à celle observée dans des cellules Ba/F3 exprimant le récepteur FGFR1 sauvage cultivées dans les même conditions (Huang et al., 1995).
FGFR1 et la voie mTOR/P70 S6Kinase:
Dans le cas du couple FGF-FGFR1, la p70 S6kinase est activée dans des cellules traitées au FGF associée à une phosphorylation de AKT (Kahan et al., 1992; Kanda et al., 1997). Cette activité p70 S6kinase est importante pour la prolifération des cellules mais n'a aucun effet sur la différenciation de cellules endothéliales traitées par le FGF. L'activité p70 S6kinase est inhibée par la wortmannine et la rapamycine, cependant aucune activité PI3Kinase n'a été détectée dans ces cellules (Kanda et al., 1997). La capacité d'inhiber l'activité de la p70 S6kinase indépendamment de la PI3kinase a également été mise en évidence par Hara et al., (1995). Des cellules surexprimant une forme dominante négative D p85 de la PI3Kinase sont toujours capables d'activer la p70 S6kinase en réponse à l'insuline.
FGFR1 et la voie des facteurs de transcription STATs:
Une voie de transduction activée par FGFR1 et impliquée dans la transformation cellulaire, est la voie des facteurs de transcription de la famille STAT.
Les protéines de la famille STAT, sont des facteurs de transcription cytosoliques activés en réponse aux cytokines et aux facteurs de croissance. Chez les mammifères, sept membres ont été identifiés: STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b et STAT6. Les protéines STATs sont présentes à l'état latent sous forme monomérique dans le cytoplasme des cellules au repos. Suite à l'hétérodimérisation du récepteur induite par la fixation du ligand, elles sont recrutées au niveau du récepteur via une interaction impliquant leur domaine SH2 et un résidu tyrosine phosphorylé au niveau du récepteur. Elles sont ensuite activées par phosphorylation sur résidu tyrosine. Cette phosphorylation permet leur dimérisation, puis leur translocation dans le noyau et leur fixation sur des séquences d'ADN spécifiques. Les protéines STATs sont impliqués dans plusieurs processus biologiques incluant la prolifération cellulaire, la différenciation cellulaire, la prévention et l'induction de l'apoptose (pour revues Bowman et al., 2000; Bromberg et al., 2001).
Peu d'études ont été menées concernant l'activation des STATs par les récepteurs FGFRs. Il est connu que FGFR1 active STAT1 et STAT3 (Hart et al., 2000) (Tableau 1), et cette activation pourrait induire des signaux de survie et de prolifération.
D'autres substrats, associés à FGFR1 activé, restent certainement à découvrir reflétant la diversité des voies de transduction. Ces protéines dialoguent principalement par des interactions moléculaires et des réactions de phosphorylation. Celles-ci ne sont pas spécifiques des FGF, puisqu'elles sont impliquées dans la transmission du signal de nombreux facteurs de croissance et neurotrophines (Schlessinger et al., 2000).
Tableau 1: Substrats connus de FGFR1 (classés du domaine juxtamembranaire au domaine extracellulaire), sites dans FGFR1 requis pour leur phosphorylation lorsqu'ils sont connus et réponses cellulaires induites.
|
Substrats de FGFR1 |
Sites dans FGFR1 |
Réponses cellulaire |
Références |
|
FRS2/SNT-1 |
- Résidus 401-434 dans le domaine juxtamembranaire - Phosphorylation sur tyrosine |
Prolifération- migration différenciation activation de la voie RAS-MAPK via la fixation à GRB2/ SOS |
Kouhara et al., 1997 Xu et al., 1998 Ong et al., 2000 Hadari et al., 2001 |
|
CRK |
- Tyrosine Y463- phosphorylée - Association et phosphorylation |
Prolifération Survie |
Larsson et al., 1999 Mayer et al., 1988 Akagi et al., 2000 Stam et al., 2001 |
|
SHC |
- Association indirecte? - phosphorylation sur tyrosine |
Survie- Prolifération- différenciation- activation de la voie RAS-MAPK via la liaison à GRB2/ SOS |
Pelicci et al., 1992 Klint et al., 1995 Gu et al., 2000 |
|
GRB2 |
- Association indirecte |
Prolifération-différenciation- activation de la voie RAS-MAPK |
Lowenstein et al., 1992 Shannon 1995 |
|
SHP2 |
- Association indirecte - Phosphorylation sur tyrosine |
Prolifération- différenciation- activation de la voie MAPK via la fixation à FRS2 |
Hadari et al., 1998 |
|
GAB1 |
- Association indirecte - Phosphorylation sur tyrosine |
Prolifération- Survie- activation de la voie PI3K via la fixation au complexe FRS2/GRB2 |
Ong et al., 2001
|
|
PI3K/AKT |
- Association indirecte via Gab1 |
Survie Synthèse protéique |
Ong et al., 2001 Browaeys-Poly& al 2000 |
|
PLC g1 |
- Tyrosine Y766- phosphorylée - Association directe et phosphorylation |
Prolifération |
Burgess et al., 1990 Mohammadi et al., 1991 |
|
STAT1/3 |
? |
Survie-Prolifération |
Hart et al., 2000 |
La dérégulation de l'activation des FGFRs peut induire des réponses mitogéniques inadaptées et participer au développement de nombreuses maladies. La transformation oncogénique peut être déclenchée par des mutations ponctuelles, une amplification génique ou des réarrangements chromosomiques.
2-4-1. Mutations ponctuelles dans les FGFRs
De nombreuses mutations ponctuelles dans les FGFRs sont impliquées dans une série de maladies héréditaires de la croissance osseuse chez l'homme (Tableau 2). Ces maladies peuvent être répertoriées en deux classes de désordres squelettiques: la craniosténose et le nanisme. Ces mutations germinales touchent les différentes parties des FGFRs comprenant la partie extracellulaire, le domaine transmembranaire et le domaine catalytique intracellulaire (Webster et al., 1997a; Kannan et al., 2000).
Des mutations ponctuelles dans la région extracellulaire des FGFRs ont été trouvées et particulièrement dans la région comprise entre la seconde et la troisième boucle immunoglobuline (Ig) (résidus 252 et 253) qui est extrêmement conservée au sein de la famille des FGFRs et qui participe à la fixation du ligand. Ces mutations sont associées à divers syndromes (pour revues Webster et al., 1997a; Burke et al., 1998) (Tableau 2). Par exemple le syndrome de Pfeiffer, une forme de craniosténose qui se caractérise par des anomalies crâniennes, est associé à une mutation d'une proline en arginine P252-R dans le segment qui relie la seconde boucle Ig à la troisième boucle dans FGFR1 (Muenke et al., 1994).
Dans le syndrome d'Apert, une forme de craniosténose encore plus marquée, les mutations affectent la même région dans FGFR2, c'est à dire la proline P253 mutée en arginine (Wilkie et al., 1995a). Ces mutations semblent augmenter l'affinité du récepteur FGFR2 pour son ligand FGF2 (Ibrahimi et al., 2001). Anderson et al., (1998) ont notamment montré que la constante de dissociation du complexe FGFR2 muté-FGF2 est diminuée. Ces mutations affectent également la spécificité du récepteur pour son ligand: dans ce cas, FGFR2 peut être activé par plusieurs FGF qui normalement ne reconnaissent pas ce récepteur (Yu et al., 2000). Cette région est également la cible de mutations dans FGFR3. Le syndrome de Crouzon, une troisième forme de craniosténose, est associé à la mutation de la proline en arginine P250-R dans FGFR3 (Bellus et al., 1996).
La mutation la plus fréquente dans FGFR2 entraîne la disparition de la cystéine C342 qui est impliquée dans la formation du pont disulfure de la boucle Ig. Cette mutation aboutit à la formation d'un récepteur FGFR2 activé en permanence (Galvin et al., 1996). La substitution de cette cystéine peut se traduire par trois types de craniosténose: le syndrome de Crouzon, le syndrome de Jackson-Weiss ou le syndrome de Pfeiffer.
Il existe environ une dizaine d'autres mutations dans FGFR2, que je ne détaillerai pas, au niveau de la troisième boucle Ig, associées à des craniosténoses. Ces mutations ont été répertoriées par Kannan et al., (2000).
Des mutations dans la région transmembranaire des FGFRs peuvent être activatrices pour les récepteurs (Tableau 2). Dans la protéine FGFR3 la mutation d'une glycine en arginine G380-R dans le domaine transmembranaire conduit à une réduction de la croissance des os longs par diminution de l'ossification endochondrale, cela se traduisant par une achondroplasie, la forme la plus fréquente du nanisme (Rousseau et al., 1994; Shiang et al., 1994). Cette mutation dans le domaine transmembranaire de FGFR3 est à la même position que la mutation dans ERBB2. Elle induit une activation du récepteur en l'absence de ligand, le degré d'activation étant directement corrélé avec la sévérité du phénotype. Cette activation est due soit à une stabilisation de la forme dimérique du récepteur (par la formation d'une liaison hydrogène dans le domaine transmembranaire entre deux récepteurs), soit à un changement de conformation dans la partie tyrosine kinase (Webster et al., 1996b; Webster et al.,1997a; Robertson et al., 2000). Il a récemment été montré que cette mutation induit un défaut dans la régulation négative des signaux de transduction associés à FGFR3 (défaut dans l'internalisation du récepteur et de sa dégradation) (Monsonego-Ornan et al., 2000).
Dans FGFR3 la substitution d'une lysine en acide glutamique K650-E dans la boucle d'activation forme un mutant gain-de-fonction retrouvé dans un syndrome de dysplasie thanatophorique (Tavormina et al., 1995). Le récepteur devient actif en absence de ligand probablement par stabilisation de la boucle d'activation sous forme "ouverte" c'est-à-dire active (Webster et al., 1996b; Naski et al., 1996). De façon intéressante, une autre mutation dans FGFR3 a récemment été décrite à la même position que celle citée précédemment, substituant une lysine en méthionine K650-M. Cette mutation induit une activation constitutive du récepteur (environ trois fois plus importante que dans FGFR3 présentant la mutation K650-E) mais aboutit à un syndrome différent désigné SADDAN pour Sévère Achondroplasie avec un Délai dans le Développement et Acanthosis Nigricans associé à des anomalies du derme (Tavormina et al., 1999).
Enfin, une mutation substituant une asparagine en lysine N540-K dans le domaine kinase de FGFR3 est associée à un syndrome d'hypochondroplasie. Cette mutation est également une mutation gain-de-fonction en destabilisant la forme "fermée" c'est-à-dire inactive du récepteur (Bellus et al., 1995).
D'autres mutations activatrices de type somatiques ont été identifiées dans les récepteurs FGFR2 et FGFR3 associées essentiellement à des carcinomes.
Les mutations dans FGFR2 affectent la partie extracellulaire du récepteur (S267-P) et sont associées à des carcinomes gastriques (Jang et al., 2001). Ces mutations sont identiques à celles retrouvées dans le syndrome de Crouzon (Tableau 2).
Les mutations dans FGFR3 sont associées à des carcinomes de la vessie et du cou (Cappellen et al., 1999). Ces mutations sont identiques aux mutations germinales responsables de la dysplasie thanatophorique. Dans les carcinomes du cou, elles sont retrouvées dans la partie extracellulaire du récepteur S249-C (Cappellen et al., 1999; Wu et al., 2000; Billerey et al., 2001; Sibley et al., 2001). Dans les carcinomes de la vessie elles touchent la partie extracellulaire (S249-C, R248-C), la partie transmembranaire (G370-C) et la partie kinase du récepteur (K670-E) (Cappellen et al., 1999).
Il a récemment été décrit une nouvelle mutation E322-K dans la troisième boucle immunoglobuline de FGFR3 dans des carcinomes colorectaux (Jang et al., 2001). Le résidu acide glutamique (E322) est très conservé dans la famille des FGFRs. La structure cristallographique de la région extracellulaire de FGFR1, complexée avec son ligand FGF2, a montré que le résidu E322 est impliqué dans l'interaction directe entre FGF2 et FGFR1 (Plonikov et al., 1999). La mutation de ce résidu doit donc empêcher l'interaction de FGF2 ce qui a pour conséquence la perte de régulation de l'activité du récepteur par le ligand.
|
FGFRs |
Mutation ponctuelle |
Partie mutée du récepteur |
Conséquence sur l'activité du récepteur |
Syndrome |
Références |
|
FGFR1 |
P252-R |
Domaine extracellulaire Entre la boucle IgII et IgIII |
Pfeiffer |
Muenke et al., 1994 |
|
|
FGFR2 |
P253-R ou S252-W |
Domaine extracellulaire Entre la boucle IgII et IgIII |
augmentation de l'affinité et perte de la spécificité du récepteur pour son ligand |
Apert |
Wilkie et al., 1995a; Anderson et al.,1998; Yu et al., 2000; Ibrahimi et al., 2001 |
|
FGFR2 |
S267-P |
Domaine extracellulaire Entre la boucle IgII et IgIII |
Crouzon |
Wilkie et al., 1995b |
|
|
FGFR2 |
C278-F |
Domaine extracellulaire Dans la 3ème boucle Ig |
Pfeiffer - Crouzon |
Meyers et al., 1996 Oldridge et al., 1995 |
|
|
FGFR2 |
C342-R,G,F,S, W,Y |
Domaine extracellulaire Dans la 3ème boucle Ig |
Perte du pont disulfure de la boucle Ig. Activation constitutive |
Crouzon- Pfeiffer Jackson-Weiss |
Park et al., 1995 Rutland et al., 1995 Galvin et al., 1996 |
|
FGFR3 |
R248-C S249-C |
Domaine extracellulaire Entre la boucle IgII et IgIII |
Pont disulfure crée Homodimérisation et activation constitutive |
(TDI) Dysplasie Thanatophorique |
Tavormina et al., 1995a et b Naski et al., 1996 |
|
FGFR3 |
P250-R |
Domaine extracellulaire Entre la boucle IgII et IgIII |
Craniosténose |
Bellus et al., 1996 |
|
|
FGFR3 |
G380-R G375-C |
Domaine transmembranaire |
activation constitutive stabilisation par formation d'une liaison H entre les dimères |
Achondroplasie |
Rousseau et al., 1994 ; Shiang et al., 1994; Webster et al., 1996a; Monsonego -Ornan et al., 2000 |
|
FGFR3 |
G370-C |
Domaine transmembranaire |
(TDI) Dysplasie Thanatophorique |
Rousseau et al., 1996 |
|
|
FGFR3 |
K650-E |
Boucle d'activation dans le domaine kinase |
activation constitutive |
(TDII) Dysplasie Thanatophorique |
Tavormina et al.,1995a; Webster et al., 1996b; Naski et al., 1996 |
|
FGFR3 |
K650-M |
Boucle d'activation dans le domaine kinase |
activation constitutive |
SADDAN |
Bellus et al., 1999 Tavormina et al.,1999 Iwata et al., 2001 |
|
FGFR3 |
N540-K I538-V |
Domaine tyrosine kinase |
activation constitutive |
Hypochondropla-sie |
Bellus et al., 1995 Grigelioniene 1998 |
Craniosténoses: Syndrome de Pfeiffer, Syndrome d'Apert, Syndrome de Crouzon, Syndrome de Jackson-Weiss.
Nanismes: Dysplasie Thanatophorique (TDI, TDII), Achondroplasie, SADDAN (Severe Achondroplasia with Developmental Delay and Acanthosis Nigricans), Hypochondroplasie.
Les diverses mutations ponctuelles, décrites précédemment et retrouvées dans certaines parties des FGFRs, activent les récepteurs de plusieurs façons:
i- par stabilisation de l'oligomérisation
ii- par modification de l'affinité du récepteur pour son ligand en diminuant la constante de dissociation
iii- par activation constitutive en stabilisant la boucle d'activation dans le domaine kinase.
L'activation du récepteur en l'absence de ligand semble être une caractéristique commune à de nombreux désordres squelettiques. La sévérité du phénotype est proportionnelle au degré d'activation de la kinase (Neilson et al., 1996; Kannan et al., 2000).
FGFR1 est amplifié dans des tumeurs de l'ovaire et des cancers du sein (Theillet et al., 1993; Ugolini et al., 1999). Cependant, son transcrit n'est pas toujours surexprimé dans les cancers du sein. FGFR1 est surexprimé dans des hyperplasies bénignes de la prostate, au niveau transcriptionnel et protéique. Dans cette étude aucune surexpression de la forme FGFR2-IIIc et de FGFR3 n'a été détectée (Boget et al., 2001).
FGFR2 est amplifié dans 5 à 10% des cancers du sein (Adnane et al., 1991; Penault-Llorca et al., 1995) et dans des lignées humaines de cancers du sein (Tannheimer et al., 2000).
FGFR4 est amplifié et surexprimé dans des carcinomes mammaires et ovariens (Jaakkola et al., 1993).
2-4-3. Réarrangements chromosomiques
Peu d'exemples montrent l'implication des FGFRs dans certaines hémopathies malignes. Seuls FGFR1, localisé sur le chromosome 8, et FGFR3, localisé sur le chromosome 4, peuvent être dérégulés suite à des translocations chromosomiques.
FGFR3 dans le myélome multiple
FGFR3 est associé à la translocation t(4;14) dans le myélome multiple, une forme de prolifération lymphoïde chronique. Cette translocation met le gène FGFR3 sous le contrôle de l'enhancer des IgH ce qui aboutit à la surexpression de FGFR3 (Chesi et al., 1997; Richelda et al., 1997). Des mutations activatrices dans FGFR3, associées au myélome multiple, ont également été mises en évidence. Une mutation dans le domaine extracellulaire de FGFR3 (R248-C), associée à la dysplasie thanatophorique (Tableau 2), est retrouvée chez des patients atteints de myélome multiple (Intini et al., 2001). Dans des lignées cellulaires portant la translocation t(4;14), des mutations dans le domaine transmembranaire Y373-C (Chesi et al., 1997; Richelda et al., 1997) et G384-D (Ronchetti et al., 2001) ainsi que dans le domaine kinase K650-E, K650-M ont été également mises en évidence (Chesi et al., 1997). Par conséquent, l'hyper-expression et une mutation activatrice dans le récepteur FGFR3 semblent donc être nécessaires pour déterminer la progression de la transformation maligne (Chesi et al., 2001).
Récemment une translocation t(4;12)(p16;p13) a été décrite dans un lymphome T associant les gènes TEL et FGFR3 (Yagasaki et al., 2001). Le gène de fusion résultant de cette translocation code pour une protéine ayant le domaine d'oligomérisation de TEL fusionné au domaine à activité tyrosine kinase de FGFR3.
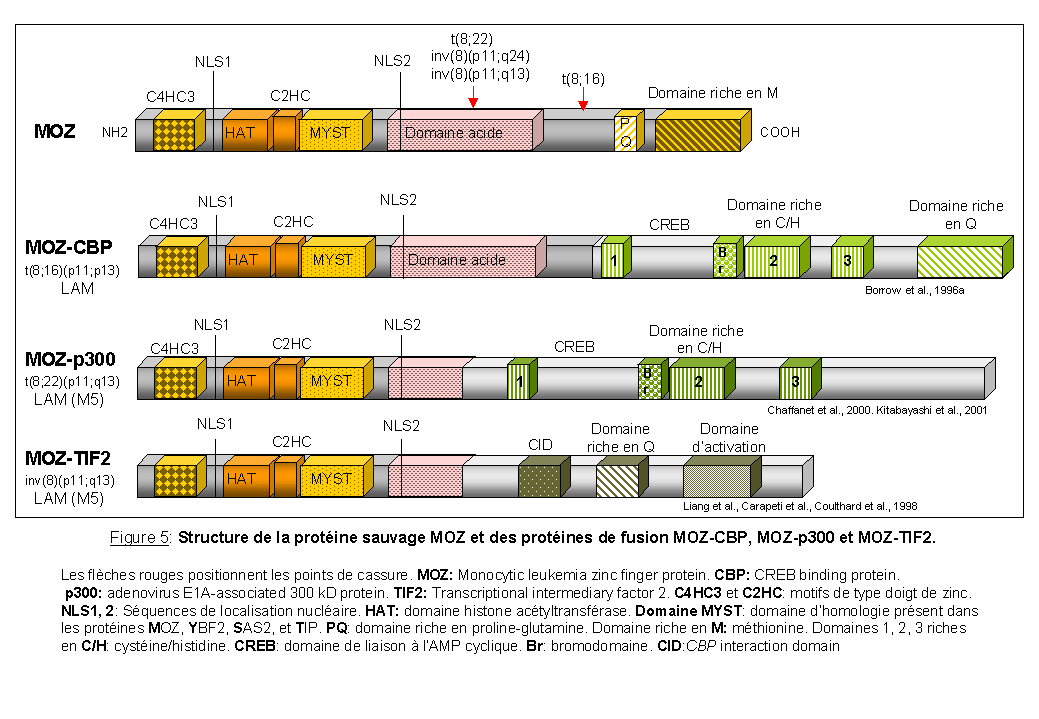
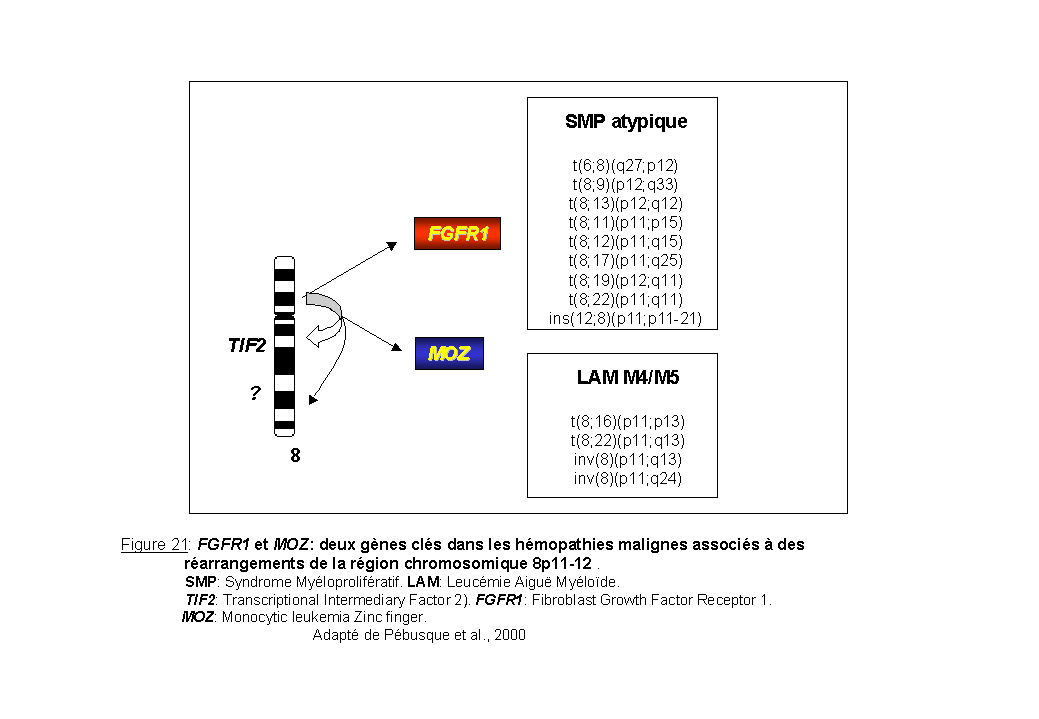
La région chromosomique 8p11-12 est le siège de plusieurs aberrations chromosomiques équilibrées (translocations et inversions péricentriques) associées à des hémopathies malignes. A l'heure actuelle, treize réarrangements chromosomiques différents ont été décrits pour cette région. Ces aberrations chromosomiques restent rares et sont associées à deux phénotypes malins: des leucémies aiguës myéloïdes de type myélomonocytaires (M4) et monocytaire (M5) et une forme de syndrome myéloprolifératif atypique (Mc Donald et al., 1995; Aguiar et al., 1997. Pébusque et al., 2000 pour revue). Les deux gènes impliqués ont été identifiés par hybridation in situ sur des chromosomes métaphasiques provenant de cellules de patients. Il s'agit respectivement de MOZ (Figure 5) (Borrow et al., 1996a) et FGFR1 (fibroblast growth factor receptor 1) (Chaffanet et al., 1998; Mugneret et al., 2000; Sohal et al., 2001) (Figure 21).
FGFR1 et le syndrome myéloprolifératif atypique 8p12
Plusieurs cas de syndrome myéloprolifératif atypique présentent comme anomalie chromosomique spécifique, un réarrangement de la région 8p11-12. Les translocations équilibrées retrouvées dans ce syndrome impliquent toujours la région p12 du bras court du chromosome 8 et le gène FGFR1. Cette région est réarrangée avec différentes régions partenaires, 6q27, 9q33, 11p15, 12q15, 13q12, 17q25, 19q13, 22q11, (Figure 21).
Le syndrome myéloprolifératif 8p12 est une hémopathie maligne rare. En effet, les cas décrits dans la littérature sont peu nombreux et sont détaillés dans le tableau 3. La translocation t(6;8) est très rare. Seuls six cas ont été décrits (tableau 3 (1),(2)). Un syndrome myéloprolifératif atypique avec éosinophilie associé à un lymphome à cellules T a été décrit chez un patient (Vannier et al., 1984). Un deuxième patient présentait uniquement un syndrome myéloprolifératif atypique avec éosinophilie (Sohal et al., 2001). Un troisième patient présentait un syndrome myéloprolifératif atypique avec éosinophilie, sans lymphome mais avec une métaplasie myéloïde dans les ganglions lymphatiques (Elsner et al., 1994).
Les hémopathies malignes associées à la translocation t(8;9) sont elles aussi rares. Neuf cas ont été décrits dans la littérature (tableau 3 (1)). L'aspect clinique est assez variable chez ces patients. La prolifération lymphocytaire chronique n'est prouvée que dans deux cas (Nakayama et al., 1996; Van den Berg et al., 1996) et pour un cas le syndrome myéloprolifératif est décrit comme une leucémie myélomonocytaire chronique (Nakayama et al., 1996).
Quinze cas de translocation t(8;13) ont été décrits (tableau 3 (2), (3)). Tous les cas sont très homogènes du point de vue clinique: un syndrome myéloprolifératif avec éosinophilie associé à un lymphome T de type non-Hodgkinien. Dans deux cas la lignée cellulaire B a été affectée sous la forme d'un lymphome B et d'une leucémie aiguë lymphoïde avec cellules B (Michaux et al., 1996).
En résumé, la majorité des patients présentent une hyperleucocytose, souvent >50.109/l, (les valeurs normales du nombre de leucocyte sont les suivantes: adulte = 4 à 9.109/l de sang; enfant de moins de 1 an = 10 à 20.109/l de sang). Cette hyperleucocytose est liée au passage dans le sang d'éléments médullaires granuleux (myélémie), une hyperplasie myéloïde granuleuse associée dans la plus part des cas à une atteinte des organes lymphoïdes périphériques sous la forme d'un lymphome T ou B non hodgkinien associé pour deux cas d'une métaplasie myéloïde (Behringer et al., 1995; Somers et al., 1997), ainsi qu'à une éosinophilie parfois très importante (McDonald et al., 1995; Inhorn et al., 1995). Le double phénotype myéloïde et lymphoïde des cellules malignes suggère que l'anomalie génétique a eu lieu dans la cellule souche hématopoïétique pluripotente ou multipotente et non au niveau d'une cellule déjà engagée dans une voie de différenciation. Le syndrome myéloprolifératif dit atypique désigne donc une prolifération chronique touchant les cellules souches hématopoïétiques médullaires ("myélo") et non uniquement de la lignée myéloïde. Chez les patients atteints de SMP 8p12, l'absence du chromosome Philadelphie a été montré grâce à des examens cytogénétiques. L'absence du transcrit BCR-ABL a été vérifiée suite à des expériences de RT-PCR à partir de cellules de moelle osseuse (tableau 4).
La comparaison du SMP 8p12 avec les différents SMP "classiques" montre des caractéristiques cliniques et biologiques communes et distinctes (tableau 4).
Le SMP 8p12 est très proche de la LMC qui est associée à la présence de la translocation chromosomique t(9;22). Ces deux maladies sont mortelles à court terme et comportent trois phases:
i- une phase chronique pendant laquelle une prolifération granuleuse maligne donne des polynucléaires neutrophiles. Une éosinophilie et une basophilie du sang périphérique sont fréquemment observées dans les deux cas. L'hématopoïèse extramédullaire dans la pulpe rouge splénique entraîne une splénomégalie importante (tableau 4). Une hépatomégalie peut dans certains cas être associée à la splénomégalie. Des lymphadénopathies sont retrouvées uniquement dans le SMP 8p12. Cependant certains patients atteint du SMP 8p12 ne présentent aucun signe clinique d'hématopoïèse extramédullaire (tableau 3 (1)), (Mugneret et al., 2000). Dans le SMP 8p12 et dans la LMC, la moelle osseuse est hypercellulaire et riche en cellules granuleuses. Le taux de blastes est normal (<5%).
ii- une phase d'accélération qui dans certains cas passe inaperçue due à l'aggressivité de la phase blastique. La phase d'accélération est caractérisée par l'apparition, la réapparition ou la majoration des signes cliniques tels que l'altération de l'état général, la fièvre, la splénomégalie. Dans la phase d'accélération une partie des cellules ne peuvent plus maturer (hyperleucocytose avec une augmentation des formes immatures). Le patient devient résistant au traitement et peut mourir de complications pendant cette phase (Jotterand et al., 1992).
iii- une phase d'acutisation (une transformation en leucémie aiguë). Dans la LMC la crise blastique peut se manifester sous forme de leucémie aiguë myéloïde ou lymphoïde (tableau 4). Les cellules blastiques contiennent fréquemment des anomalies chromosomiques additionnelles au chromosome Philadelphie, dont les plus fréquentes sont un deuxième chromosome Philadelphie et une trisomie 8. Chez les patients atteints du SMP 8p12 qui n'ont pas bénéficié de transplantation médullaire, après une phase chronique d'environ 9 mois, le syndrome myéloprolifératif atypique évolue vers une transformation leucémique myéloïde aiguë fatale, malgré un traitement de chimiothérapie. Lors de l'entrée en phase aiguë, des clones cellulaires avec des anomalies secondaires variées peuvent apparaître dont parfois les trisomies 8 et 21, et la duplication de certains chromosomes dérivatifs tels que le der(6), der(8) et der(13) (Naeem et al., 1995; Nakayama et al., 1996; Popovici et al., 1998).
Parmi les patients qui ont bénéficié d'une transplantation médullaire, un patient présentant une translocation t(8;9) a survécu deux ans après la greffe (Van den Berg et al., 1996), un autre patient ayant une translocation t(8;13) a survécu 9 mois après ce traitement (Behringer et al., 1995). Trois patients présentant une translocation t(8;13) et ayant reçu une transplantation de moelle osseuse allogénique, sont restés en rémission 3, 6 et 26 mois après la greffe (Inhorn et al., 1995; Michaux et al., 1996; Martinez et al., 1998). Enfin nous avons récemment décrit un patient présentant une translocation t(8;13) qui se trouve en rémission complète (hématologique, cytologique et moléculaire) deux ans après une transplantation médullaire (Article n°1; tableau 3 (2)). La greffe de moelle osseuse allogénique semble donc être le meilleur traitement à envisager dans le cas du SMP 8p12 après cytoréduction.
Dès 1995, MacDonald et al., avaient émis l'hypothèse de l'existence d'un gène unique réarrangé dans les cas de syndrome myéloprolifératif présentant les translocations t(6;8), t(8;9) et t(8;13) (les six autres translocations chromosomiques n'ayant pas été mises en évidence à l'époque). Cette hypothèse a été confirmée par des travaux réalisés dans notre laboratoire montrant l'implication du locus FGFR1, localisé dans la région 8p11-12, dans ces trois événements de translocation (Chaffanet et al., 1998).
3. Références bibliographiques
BUT DU PROJET DE RECHERCHE
L'objectif de mon projet de thèse a consisté à établir les caractéristiques moléculaires du syndrome myéloprolifératif atypique, lié à la région chromosomique 8p12. La première étape a été de cloner les gènes de fusion impliqués dans le syndrome malin, puis de caractériser les altérations dans leur fonction et les processus cellulaires mis en jeu. Ce travail s'est poursuivi par la mise en place d'un modèle expérimental dans lequel nous avons tenté de reproduire la maladie chez des souris greffées avec des cellules hématopoïétiques exprimant les protéines de fusion.
Ce chapitre est issu de la THESE de Doctorat de Géraldine Guasch, 06 mars 2002, Université d'Aix-Marseille II
"Caractérisation moléculaire du syndrome myéloprolifératif 8p12 impliquant le gène FGFR1"
Directeur de Thèse : Mme le Docteur Marie-Josèphe Pébusque
© Copyright Université Aix-Marseille II 2002
Created: 29/04/2002
Last updated: Friday, 06-Feb-2026 15:23:24 CET
Author: Géraldine Guasch
Editors: Chantal Ginestoux and Marie-Paule Lefranc
IMGT Home page |
IMGT Repertoire (IG and TR) |
IMGT Repertoire (MH) |
IMGT Repertoire (RPI) |
IMGT Index |
IMGT Scientific chart |
IMGT Education |
IMGT Latest news ![]()



© Copyright 1995-2026 IMGT®, the international ImMunoGeneTics information system® | Terms of use | About us | Contact us | Citing IMGT
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}