Recent evolutionary advances in sequencing mean that many species can be sequenced quickly, cheaply and with long reads[1]. The number of assemblies is constantly increasing and many assemblies are subsequently reassembled or undergo major modifications. Some strains and isolates can be assembled several times. These developments represent a major challenge for IMGT, as the nomenclature of IG and TR genes is based on generated assemblies. The amount of data highlights the allelic and genetic diversity between different individuals, but it is difficult to keep track of all the published assemblies. In addition, these technological advances also pose challenges in terms of assembly quality. Poor reconstruction or sequencing errors can lead to incorrect gene nomenclature and allele naming. Recognizing its role in the analysis and description of IG and TR genes of jawed vertebrate (Gnathostomata) and its position as the internationally recognized authority in this field, IMGT has decided to implement quality measures to mitigate the risks of annotating and including assemblies with limited reconstruction or nucleotide certainty within the IG and TR loci. This methodology involves locus analysis on the assembly and re-analysis of the reads used for assembly, focusing on the loci of interest and the exonic regions of the of the IG and TR genes.
1. What data are used to identify genes and alleles?
In most cases, our assemblies come from general databases such as INSDC databases (NCBI or EMBL-EBI), NGDC databases or other reputable databases. We may also receive data from collaborators. In this case, only public data will be published on the IMGT website. Additionally, we will consider annotating single or multiple genes if the scientific evidence allows us to describe them with confidence. Priority will be given to species with significant scientific impact.
2. IMGT nomenclature for IG/TR genes and alleles
IMGT nomenclature is based on a set of rules and can be separated in two groups:
- Definitive nomenclature
- The gene is given a definitive name when its position and existence on the locus can be identified with certainty.
- Provisional nomenclature
- The gene is given a provisional name when its existence or position cannot be guaranteed.
3. Analysis of input assemblies
Based on this analysis, which would be made available upon request, we would decide whether to issue a final IMGT nomenclature[4] or a provisional one. If the quality does not meet our standards, we could also decide not to validate any nomenclature.Global analysis of assembly and loci
The following criteria are used to evaluate an assembly:- Haploid
- A haploid assembly is preferred for allele identification. Therefore, an assembly where both haplotypes are available would be ideal for identifying genetic variation.
- Number of assemblies
- To compare individuals and ensure consistency, the number of available assemblies is taken into account.
- Recency
- A recent assembly would be preferred if its reconstruction, sequencing method, or assembly methodology is considered superior to that of an older one.
- Assembly level
- A chromosome-level assembly is preferred as it provides more information about the locus.
- Completeness of the locus
- An assembly with the expected number and organisation of V/D/J/C and RPI genes, with no nucleotide gaps or vector contamination and identified bornes would be preferred as confidence in locus position and gene order would be higher. A locus identified far from sequence boundaries (if possible) is also preferred for completeness. On the contrary, a rearranged locus (from B/T cells for instance) would be avoided.
- Litterature
- The selection of assemblies is also based on literature and international reputation.
- Assembly construction
- The sequencing method (PacBio, Oxford Nanopore, Illumina...) as well as the assembly method (Hifasm, Flye, Vekko...)[2] are considered. An assembly with HiFi long reads, a good coverage and a haplotype-resolved assembly method[3] would be preferred. Some assembly methods have already been compared for the overall assembly quality [7].
- Assembly analysis (see section below)
Reads analysis
To analyse assembly reads, we will extract the reads from the
assembly and perform a global analysis on the reads, preferably
HiFi reads(1).
The software we
will use is called IMGT/StatAssembly[8,9](2).
It uses the result of a BAM
alignment file mainly provided by minimap2[5].
Graphical and textual outputs are provided, allowing comparison of the generated assembly with the underlying HiFi reads to assess confidence.
The presence of an MD/CS tag or a CIGAR =/X partition is required for full results. Partial results could be obtained without it though.
For example, with
HiFi reads in FASTQ
format, a BAM file could be generated from the reads (reads.fq)
and the assembly downloaded as a FASTA file
(assembly.fasta):
minimap2 -t 28 --cs --eqx -ax map-hifi assembly.fasta reads.fq > align.sam
samtools sort -@ 28 align.sam -o align.bam
samtools index -c -@ 28 align.bam
samtools calmd -b -@ 28 full.bam assembly.fasta > align.bam
The generated alignment file is then analysed with IMGT/StatAssembly, depending on the reads. Here is an example (more details on Gitlab):
IMGT_StatAssembly -f align.bam -l locus.txt -s human -o output_dir/
Results of analysis - human PGPv1
Results are shown for the assembly human assembly PGPv1 with IMGT/StatAssembly (version 0.1.3):
Global assembly analysis
- A haploid assembly
- There are hundreds of assemblies for human
- The assembly is from 2011
- This is a chromosome-level assembly
- The assembly contains the expected structure of the human IG locus
- It is used in the literature to compare assemblies
- It was constructed using Illumina/ONT/Pacbio Hifi reads with the haplotype-unaware Flye assembler.
Read analysis
Assembly quality
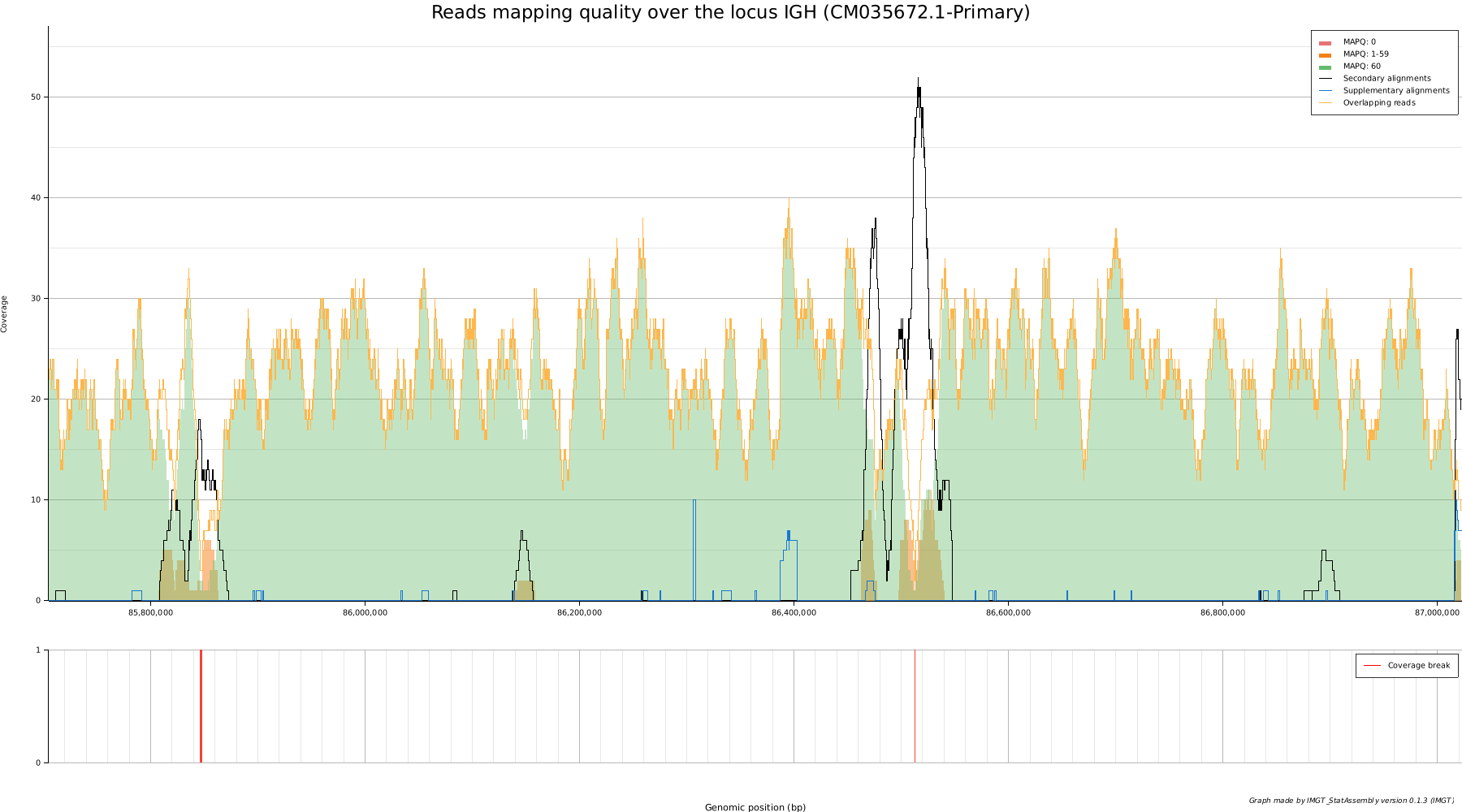
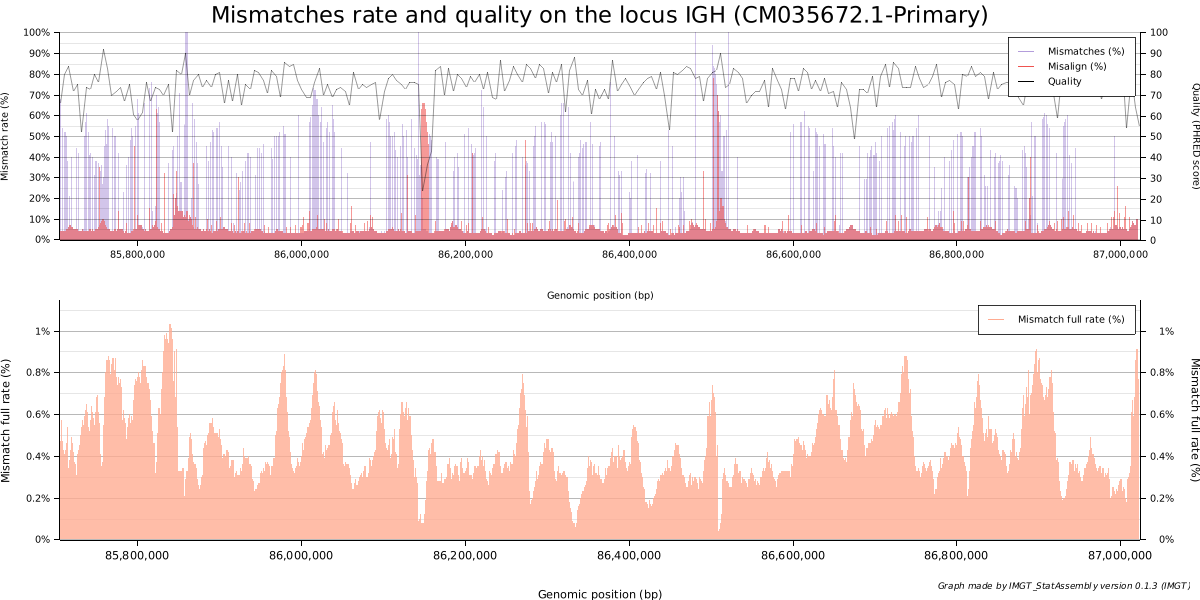
A good quality assembly would meet most of the criteria:
- A good and uniform coverage (green curves)
- A negligible fraction of secondary and supplementary alignments (in black and blue)
- No breaks (in red), where reads overlap insufficiently
- Quality reads Q>30 (PHRED quality score)
- A negligible amount of mismatches and misalignments.
- Low overall mismatch rate for reads
In this case, we can see two gaps and two regions of uncertainty that could be analyzed and compared to other assemblies. These areas are low-complexity regions (LCR), and poor mapping quality is to be expected. Some regions also have a high rate of misalignment and mismatch.
Allele quality
Alleles could also be checked if their positions are known and passed to the script. In this scenario, the following graphs are generated:
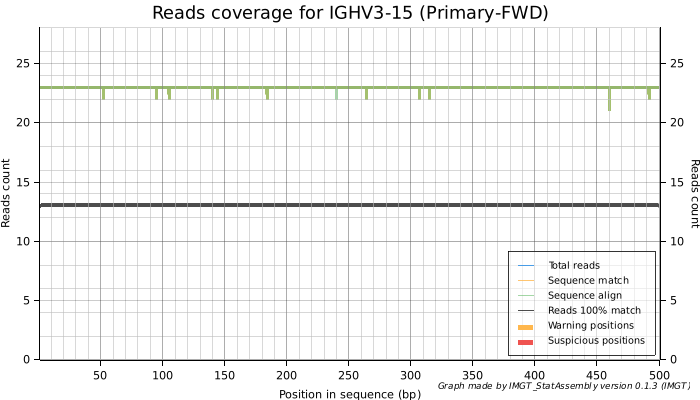

A good quality allele would fulfil most of the criteria:
- No warning or suspicious positions.
- Reads that match the exonic regions of the gene with 100% accuracy (black curve). A higher ratio compared to the total reads is preferred.
- An average coverage close to or greater than 100%.
We can see here that IGHV3-15, named IGHV3-15*01 seems to be validated by these reads, it is also found in many assemblies. However IGHV4-4, named IGHV4-4*12, seems to have low-confidence, is not found in other assemblies and is probably an artefact of sequencing errors. It has therefore been removed from IMGT databases (December 17th, 2024).
4. What data are published?
After the validation of assemblies and alleles, as well as a detailed and meticulous annotation[6] and study of genes within a locus, the locus annotation and its genes/alleles are published in the IMGT databases and on the website. Data are be announced through the creations and updates tab and the RSS news feed. Additionally, annotated assemblies are published in INSDC databases.Annotations are available in LIGM-DB. Genes and alleles are also deposited in GENE-DB databases and accessible through tools such as IMGT/V-QUEST and HighV-QUEST. Visual representations of the data are available on IMGT Repertoire.